球脊髄性筋萎縮症におけるSrc病態
名古屋大学大学院医学系研究科 神経内科学
発行日:2021年6月30日Published: June 30, 2021
© 2021 日本神経化学会© 2021 The Japanese Society for Neurochemistry
運動ニューロンは骨格筋を支配している神経細胞であり、細胞体は主に大脳皮質の運動野と脊髄前角に存在する。運動ニューロン疾患は運動ニューロンの選択的変性・脱落により、進行性の筋力低下や筋萎縮をきたす。成人発症の運動ニューロン疾患の大多数は筋萎縮性側索硬化症(ALS)と球脊髄性筋萎縮症(SBMA)であり、いずれの疾患も主に呼吸不全により死に至る。運動ニューロン疾患の分子病態は未だ解明されていない点が多く、根本的治療法は見出されていない。SBMAは運動ニューロンと骨格筋の変性・脱落をきたす遺伝性の難病である。2017年にリュープロレリン酢酸塩が治療薬として承認されたが、副作用や効果が限定的であることからさらなる治療薬の開発が望まれている。SBMAの病態、治療法開発の過程、さらに今後の治療の可能性についてわれわれの最新の研究結果を交えて概説する。
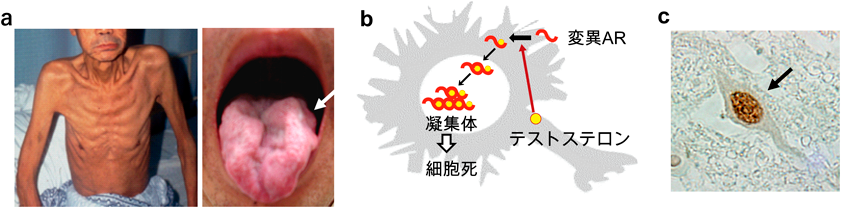
SBMAは男性のみに発症する遺伝性の神経筋疾患であり、緩徐に進行する筋力低下や筋萎縮をみとめる。筋症状は四肢近位や顔面、構音筋にみられ、歩行障害や話しにくさ、飲み込みにくさ、鼻声を呈する1)(図1a)。日本の有病率は人口10万人あたり2人程度であり、人種や地域による差は明らかでない。手指の振戦や有痛性の筋痙攣が先行することが多く、30~60歳で筋力低下を初めて自覚する。下肢遠位優位に振動覚低下をみとめることがあり、随伴症状として女性化乳房、発毛の減少、睾丸萎縮などのアンドロゲン不応症状がみられる。知能や精神は正常であり、嚥下・呼吸機能低下による呼吸器感染が死因になることが多い。
血液検査では、筋細胞の障害を反映して血清クレアチンキナーゼ(CK)が上昇(正常値~正常上限の10倍以上)する。血清クレアチニンは筋萎縮を反映し低値であり、運動機能などの重症度を反映するバイオマーカーとなる可能性がある2)。また肝機能障害や脂質代謝異常、耐糖能異常を伴う。内分泌学的検査ではアンドロゲン抵抗性がみられるが、血清テストステロンは正常ないし軽度高値である。針筋電図検査(細い電極針を筋肉に刺して筋線維の電気活動を調べる)では、進行性かつ慢性の脱神経所見がみられる。神経伝導検査(末梢神経を電気刺激して刺激が伝わる速さや程度を検査する)では、特に感覚神経である腓腹神経において活動電位の低下をみとめる3)。心電図では約10%でBrugada型異常がみられ、一部の患者で失神や突然死に至ることがある。嚥下造影では喉頭蓋谷や食道の入口にバリウム残留をみとめ、嚥下機能の評価に用いる。病理学的には脊髄前角、顔面神経核、舌下神経核、疑核のニューロンの変性・脱落をみとめ、骨格筋では神経原性変化(運動ニューロンが障害されて起こる変化)と筋原性変化(筋細胞が障害されて起こる変化)が混在する。2010年から遺伝子検査に保険が適応となり、診断に有用である。
SBMAの病因はアンドロゲン受容体(AR)遺伝子のCAG反復配列の異常伸長である。ARはX染色体長腕(Xq11-q12)上の第1エクソンに存在し、健常人ではCAGリピート数が11~36であるがSBMA患者では39~72である。リピート数が多いほど若年発症になるが、CAGリピート数と症状の進行速度は関連がないと知られている。グルタミンをコードするCAG塩基配列が原因遺伝子において異常に伸長する疾患は、ポリグルタミン病と総称される。ポリグルタミン病は、脊髄小脳変性症やハンチントン病など現在9つの疾患が知られている。これらの疾患では神経細胞内に変異蛋白が蓄積する。異常な折りたたみ構造を呈する変異蛋白が不溶性のオリゴマーを形成してニューロンの核内に集積し、転写障害やDNA損傷、軸索輸送障害などの細胞の機能低下を引き起こす可能性が示唆されている。ARは生殖器の他、中枢神経、心臓、骨格筋、腎、副腎、膵臓など多くの臓器に発現しており、多彩な症状に反映している。
SBMAの原因蛋白質であるARは核内受容体であり、熱ショック蛋白質(HSP)などと複合体を形成し細胞質に存在するが、リガンドである男性ホルモン(テストステロン)と結合するとこれらの蛋白質から離れて核内へ移行し、転写を制御する。変異ARは核内に移行すると凝集体を形成し、細胞死を引き起こす(図1b, c)。ARのCAGリピートが97個に伸長したSBMAマウスモデル(AR-97Qマウス)では雄のみに進行性の筋力低下や神経原性筋萎縮をみとめ、患者と同様の症状を呈する。一方去勢術を行い、男性ホルモンを低下させると、運動ニューロンなどにおける変異ARの集積が著明に減少し、表現型や寿命も改善する4)。またAR-97Qマウスに、テストステロン分泌を抑制するリュープロレリン酢酸塩を投与すると病態抑止効果をみとめる5)。一方雌マウスにテストステロンを投与すると、変異ARの核内集積が増加し筋力低下や筋萎縮が生じる。以上より変異ARがテストステロンの存在により核内に集積し、細胞障害をきたすことがSBMAの病態であると考えられている。
上述の研究結果に基づいて行われた、SBMA患者50例に対する第II相臨床試験(ランダム化プラセボ対照化比較試験)では、リュープロレリン酢酸塩の投与により陰嚢皮膚における変異ARの核内集積の抑制、血清CKの改善が明らかになった6)。その後の第III相臨床試験では、リュープロレリン酢酸塩投与群100名、プラセボ投与群99名の計199名の患者に投薬が行われた。主要評価項目である咽頭部バリウム残留率の48週間の変化量はリュープロレリン酢酸塩群で−5.1%、プラセボ群で0.2%であった(p=0.063)。また発症10年未満の群におけるサブグループ解析を行うと、咽頭部バリウム残留率の変化量がリュープロレリン酢酸塩群で−6.4%、プラセボ群で3.4%となり有意に改善した(p=0.009)。陰嚢皮膚における変異ARの核内凝集の頻度や血清CKにおいても、治療群で有意に低下していた。以上の結果より48週間のリュープロレリン酢酸塩投与は、発症からの期間が短い患者において嚥下機能を改善する可能性が考えられ7)、2017年にリュープロレリン酢酸塩がSBMAの進行抑制に関して効能追加承認を取得した。しかし、四肢筋力低下に対する効果が限定的であること、性機能抑制や骨格筋のタンパク同化作用抑制などの副作用があることから、新たな治療薬の開発が望まれている。
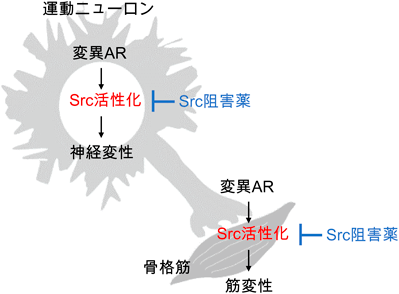
SBMAではグリアや骨格筋など非神経細胞の動態が深く関与していることが明らかになってきている。中でも運動ニューロンと骨格筋のクロストークの異常(図2)が注目されている。SBMA患者では血清CK値が上昇し、筋病理で筋原性変化をみとめることから、骨格筋における一次性の病態が示唆されている。SBMAのノックインマウスモデルでは、骨格筋変性が運動ニューロン障害に先行する8)。また変異ARが惹起するミトコンドリア異常や酸化ストレス、NFκBシグナル異常による細胞障害は、ニューロンだけでなく骨格筋においてもみられることをわれわれが報告している9)。さらにマウスの筋のみに変異ARを発現させると運動ニューロン障害を引き起こすことが明らかになっており10)、逆に骨格筋のみで変異ARの発現を抑制するとSBMAモデルマウスの病態が改善することも知られている。クレアチンは骨格筋のエネルギー代謝に重要であるが、SBMAではクレアチントランスポーターであるSLC6A8の発現低下により骨格筋へのクレアチン取込みが低下していることが報告されている11)。骨格筋は栄養因子を供給する主な組織であるが、骨格筋障害に起因するグリア細胞株由来神経栄養因子、血管内皮増殖因子などの減少が、ニューロンの機能低下の一因となっている可能性が考えられている。こうしたことから、SBMAでは運動ニューロン変性と並行して、骨格筋においても変異ARによる一次的な病態が存在することが示唆されている。

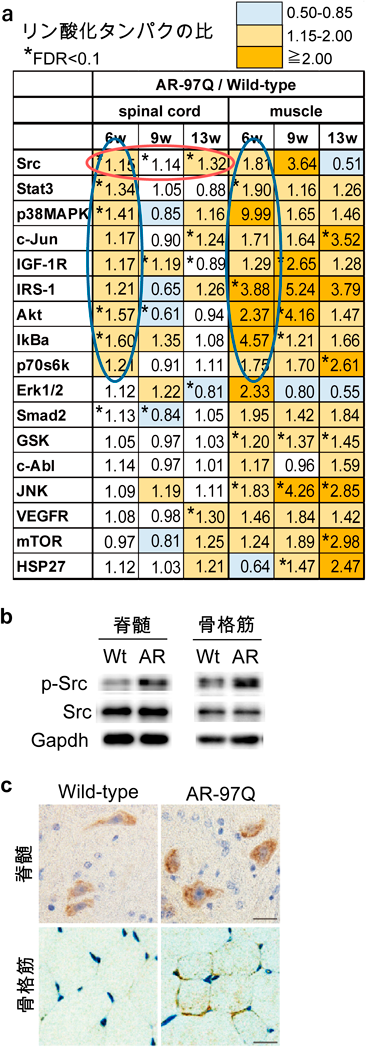
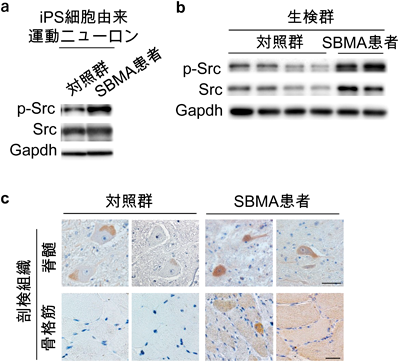
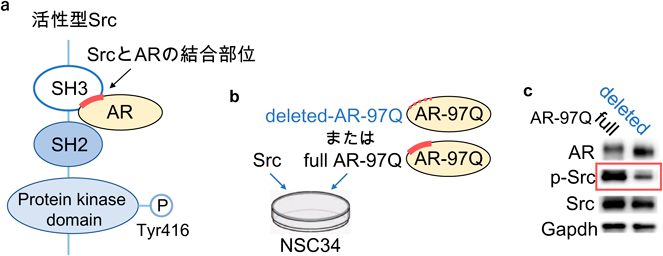
神経疾患では表現型が出現する以前に分子病態が大きく進行していることが明らかになってきている。また前述のように、SBMAでは運動ニューロンだけでなく骨格筋も治療のターゲットになりうることが示唆されている。われわれはSBMAの病態の本質に寄与する分子機序を明らかにし、より根本的な治療法を見出すことを目標として、AR-97Qマウスの脊髄と骨格筋のシグナル変化を運動機能障害が出現する以前から経時的かつ網羅的に解析した。まず、AR-97Qマウスの3つの病期(発症前、発症前期、発症後期)における脊髄と骨格筋からタンパクを抽出し、Bio-Plex™マルチプレックスシステムを用いて、主に生存や増殖に関わる様々なシグナルにおける代表的な分子(計17個)のリン酸化レベルを同時に測定した。野生型マウスと比較して、AR-97Qマウスの脊髄においてSrcのリン酸化が発症前(6週齢)から発症後期(13週齢)まで一貫して上昇し、さらにSrcの下流に存在するStat3のリン酸化が発症前に脊髄と骨格筋で上昇していた(図3)。マウスの神経幹細胞であるNSC34と、マウスの筋芽細胞であるC2C12を用いて作成したSBMA細胞モデル(AR-97Q)においてもSrcの活性化を認めた。またSBMA患者より樹立したiPS細胞由来運動ニューロン、SBMA患者の生検筋、ヒト剖検組織(脊髄と骨格筋)においてもSrcのリン酸化が上昇していた(図4)。これらの結果からSrcシグナルの異常活性化がSBMAの病態に強く関連している可能性が考えられた。
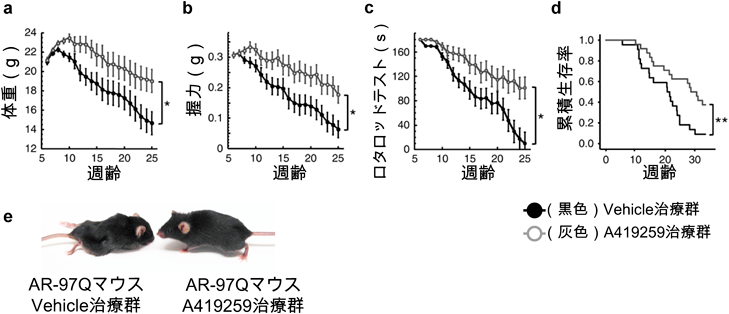
SBMA細胞モデルにSrc阻害薬(A419259)を添加することにより生存率が改善し、SBMA細胞モデルにSrcを過剰に発現させると細胞の活性が低下したため、Srcの活性化がSBMAの病態悪化に寄与していることが示唆された。またSBMAマウスモデルにA419259を腹腔内投与すると病態の抑止効果を認めた12)(図5)。A419259の投与は、SBMAマウスモデルの脊髄と骨格筋におけるARの発現量に影響を与えていなかった。Src阻害薬の作用機序を明らかにすべく、Src阻害薬投与群と非投与群のSBMA細胞モデル(神経細胞と筋芽細胞)とマウスモデル(脊髄と骨格筋)を用いてSrcのeffector moleculeである、p130Cas・Stat3・p38MAPK・Aktのリン酸化レベルについてウエスタンブロットを用いて評価した。その結果、Src阻害薬の投与によりp130Casのリン酸化が全てのモデルで有意に抑制されており、p130Casの活性化が病態に関与していることが示唆された。そこでSBMA細胞モデルにp130Casを一過性強制発現させると細胞の生存率は低下し、siRNAを用いてp130Casの発現を抑制すると細胞の活性が改善したため、p130Casのリン酸化がSBMAの病態に関与していることが明らかになった。その後われわれはSBMAにおいてSrcシグナルが活性化するメカニズムを調べた。前立腺がんの細胞モデルにおいてARとSrcが結合し相互に活性化させるという報告を基に13, 14)、NSC34にdeleted AR-97Q(ARとSrcの結合部位を欠損させたプラスミド)とSrcを一過性強制発現させると、full AR-97Q(ARとSrcの結合部位を欠損させていないプラスミド)とSrcを発現させた場合と比較してSrcの活性化が低下していることが明らかになった(図6)。これはSrcの活性化にはARとSrcの直接的な結合が重要であることを示唆する結果であった12)。
ALSは成人発症の運動ニューロン疾患であり、日本における有病率は1万人に1人である。約10%が家族性であり、その中の20%がSOD1変異を伴う家族性ALSである。われわれは、ALSのマウスモデルであるG93A変異SOD1マウスの脊髄と骨格筋を用いたBio-Plex™マルチプレックスシステムの解析を行った。その結果SBMAマウスモデルと同様に、発症前(9週齢)において脊髄と骨格筋でSrcのリン酸化が上昇していた。SOD1変異のあるALS患者由来iPS細胞から作成した脊髄運動ニューロンでは、Src/c-Ablシグナルの活性化が報告されている15)。また様々な変異を含むALS患者由来iPS細胞を用いて細胞死を抑制する薬剤をスクリーニングした研究では、Src/c-Abl阻害薬の1つであるボスチニブが運動神経細胞死と異常タンパクの蓄積を抑制することが見出されている15)。なおSrcと共に非受容体型チロシンキナーゼとして知られるc-Ablの阻害薬はG93A変異SOD1マウスの生存率を改善させたと報告されている16)。以上の知見より、Srcシグナル異常は運動ニューロン疾患に共通した病態である可能性が考えられる。
がんは細胞増殖が異常に進んでしまう疾患である。一方、運動ニューロンは通常増殖せず、運動ニューロン疾患に罹患すると細胞死が一方向性に進行することから、がんと運動ニューロン疾患は一見、逆の病態であると考えられる。今回注目したSrcは、種々の癌で活性化していることが知られており、癌の進行や転移に関連している。運動ニューロン疾患におけるSrc病態は、神経筋疾患と癌の共通項であるとともに、Srcシグナルを阻害する抗がん剤が神経難病治療薬となり得る新たな可能性が示唆された。
SBMAの新規治療薬としてSrc阻害薬が有望であることが示唆された(図7)。Src阻害薬の中には、癌の治療薬として臨床応用されている薬剤もしくは治験中の薬剤が複数存在する。SBMAの病態改善におけるそれらの薬剤の有効性が期待される。
本稿で紹介した研究におきまして、多くのご指導とご支援を頂きました名古屋大学大学院医学系研究科神経内科学の勝野雅央教授、共同研究者の方々に深く感謝申し上げます。また、今回執筆の機会を与えてくださいました神経化学会出版・広報委員会、関係者の皆様に感謝致します。
1) Sobue G, Hashizume Y, Mukai E, Hirayama M, Mitsuma T, Takahashi A. X-linked recessive bulbospinal neuronopathy. A clinicopathological study. Brain, 112(1), 209–232 (1989).
2) Hashizume A, Katsuno M, Banno H, Suzuki K, Suga N, Mano T, Atsuta N, Oe H, Watanabe H, Tanaka F, Sobue G. Longitudinal changes of outcome measures in spinal and bulbar muscular atrophy. Brain, 135(9), 2838–2848 (2012).
3) Suzuki K, Katsuno M, Banno H, Takeuchi Y, Atsuta N, Ito M, Watanabe H, Yamashita F, Hori N, Nakamura T, Hirayama M, Tanaka F, Sobue G. CAG repeat size correlates to electrophysiological motor and sensory phenotypes in SBMA. Brain, 131(1), 229–239 (2008).
4) Katsuno M, Adachi H, Kume A, Li M, Nakagomi Y, Niwa H, Sang C, Kobayashi Y, Doyu M, Sobue G. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron, 35(5), 843–854 (2002).
5) Katsuno M, Adachi H, Doyu M, Minamiyama M, Sang C, Kobayashi Y, Inukai A, Sobue G. Leuprorelin rescues polyglutamine-dependent phenotypes in a transgenic mouse model of spinal and bulbar muscular atrophy. Nat Med, 9(6), 768–773 (2003).
6) Banno H, Katsuno M, Suzuki K, Takeuchi Y, Kawashima M, Suga N, Takamori M, Ito M, Nakamura T, Matsuo K, Yamada S, Oki Y, Adachi H, Minamiyama M, Waza M, Atsuta N, Watanabe H, Fujimoto Y, Nakashima T, Tanaka F, Doyu M, Sobue G. Phase 2 trial of leuprorelin in patients with spinal and bulbar muscular atrophy. Ann Neurol, 65(2), 140–150 (2009).
7) Katsuno M, Banno H, Suzuki K, Takeuchi Y, Kawashima M, Yabe I, Sasaki H, Aoki M, Morita M, Nakano I, Kanai K, Ito S, Ishikawa K, Mizusawa H, Yamamoto T, Tsuji S, Hasegawa K, Shimohata T, Nishizawa M, Miyajima H, Kanda F, Watanabe Y, Nakashima K, Tsujino A, Yamashita T, Uchino M, Fujimoto Y, Tanaka F, Sobue G; Japan SBMA Interventional Trial for TAP-144-SR (JASMITT) study group. Efficacy and safety of leuprorelin in patients with spinal and bulbar muscular atrophy (JASMITT study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol, 9(9), 875–884 (2010).
8) Yu Z, Dadgar N, Albertelli M, Gruis K, Jordan C, Robins DM, Lieberman AP. Androgen-dependent pathology demonstrates myopathic contribution to the Kennedy disease phenotype in a mouse knock-in model. J Clin Invest, 116(10), 2663–2672 (2006).
9) Iida M, Katsuno M, Nakatsuji H, Adachi H, Kondo N, Miyazaki Y, Tohnai G, Ikenaka K, Watanabe H, Yamamoto M, Kishida K, Sobue G. Pioglitazone suppresses neuronal and muscular degeneration caused by polyglutamine-expanded androgen receptors. Hum Mol Genet, 24(2), 314–329 (2015).
10) Cortes CJ, Ling SC, Guo LT, Hung G, Tsunemi T, Ly L, Tokunaga S, Lopez E, Sopher BL, Bennett CF, Shelton GD, Cleveland DW, La Spada AR. Muscle expression of mutant androgen receptor accounts for systemic and motor neuron disease phenotypes in spinal and bulbar muscular atrophy. Neuron, 82(2), 295–307 (2014).
11) Hijikata Y, Katsuno M, Suzuki K, Hashizume A, Araki A, Yamada S, Inagaki T, Iida M, Noda S, Nakanishi H, Banno H, Mano T, Hirakawa A, Adachi H, Watanabe H, Yamamoto M, Sobue G. Impaired muscle uptale of creatine in spinal and bulbar muscular atrophy. Ann Clin Transl Neurol, 3(7), 537–546 (2016).
12) Iida M, Sahashi K, Kondo N, Nakatsuji H, Tohnai G, Tsutsumi Y, Noda S, Murakami A, Onodera K, Okada Y, Nakatochi M, Tsukagoshi Okabe Y, Shimizu S, Mizuno M, Adachi H, Okano H, Sobue G, Katsuno M. Src inhibition attenuates polyglutamine-mediated neuromuscular degeneration in spinal and bulbar muscular atrophy. Nat Commun, 10(1), 4262 (2019).
13) Zarif JC, Lamb LE, Schulz VV, Nollet EA, Miranti CK. Androgen receptor non-nuclear regulation of prostate cancer cell invasion mediated by Src and matriptase. Oncotarget, 6(9), 6862–6876 (2015).
14) Asim M, Siddiqui IA, Hafeez BB, Baniahmad A, Mukhtar H. Src kinase potentiates androgen receptor transactivation function and invasion of androgen-independent prostate cancer C4-2 cells. Oncogene, 27(25), 3596–3604 (2008).
15) Imamura K, Izumi Y, Watanabe A, Tsukita K, Woltjen K, Yamamoto T, Hotta A, Kondo T, Kitaoka S, Ohta A, Tanaka A, Watanabe D, Morita M, Takuma H, Tamaoka A, Kunath T, Wray S, Furuya H, Era T, Makioka K, Okamoto K, Fujisawa T, Nishitoh H, Homma K, Ichijo H, Julien JP, Obata N, Hosokawa M, Akiyama H, Kaneko S, Ayaki T, Ito H, Kaji R, Takahashi R, Yamanaka S, Inoue H. The Src/c-Abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Sci Transl Med, 9(391), eaaf3962 (2017).
16) Katsumata R, Ishigaki S, Katsuno M, Kawai K, Sone J, Huang Z, Adachi H, Tanaka F, Urano F, Sobue G. c-Abl inhibition delays motor neuron degeneration in the G93A mouse, an animal model of amyotrophic lateral sclerosis. PLoS One, 7(9), e46185 (2012).

This page was created on 2021-05-31T15:57:26.478+09:00
This page was last modified on 2021-07-05T13:32:48.000+09:00
このサイトは(株)国際文献社によって運用されています。