末梢神経機能に関与する翻訳リードスルータンパク質Large myelin protein zeroの解析
島根大学医学部解剖学講座
発行日:2021年6月30日Published: June 30, 2021
© 2021 日本神経化学会© 2021 The Japanese Society for Neurochemistry
ウイルス、細菌、ショウジョウバエなどの下等生物からヒトを含む高等生物までの遺伝子発現において、転写、翻訳、翻訳後などの様々な段階で調節や修飾などが行われ、1つの遺伝子から多様性を生んでいることが知られている。
その中で翻訳過程制御の一つである翻訳リードスルー(翻訳時のストップコドン・リードスルー)は下等生物など遺伝子数の少ない動物においてもタンパク質の多様性を生み出す重要なシステムである。これはmRNA本来の正統なストップコドンのリードスルー(読み飛ばし)により、読み枠を維持したまま次のストップコドンまでタンパク合成が進むメカニズムであり、新たな機能ドメインの付加などタンパク質の多様性に貢献する1, 2)(図1)。高等動物においても同様のシステムが存在すると予想されていたが、近年まで普遍的な存在は確認されていなかった。我々のグループはヒトを含む高等動物において、上記の翻訳リードスルーのメカニズムにより産生される分子としてMyelin protein zero(P0もしくはMPZ)の新しいアイソフォームであるLarge Myelin Protein Zero(L-MPZ)を見出した3)。このL-MPZの発見を皮切りに、現在までに、血管新生を促進するvascular endothelial growth factor A(VEGF-A)に対するVEGF-Ax4)および神経系のアストロサイトに発現し脳内の水分量を調節する水チャネルであるaquaporin 4(AQP4)に対するAQP4exの存在が知られている5)。しかし、これらのリードスルーによって産生されるタンパク質の機能などの多くはまだ完全に解明されておらず、いまだ不透明のままである。
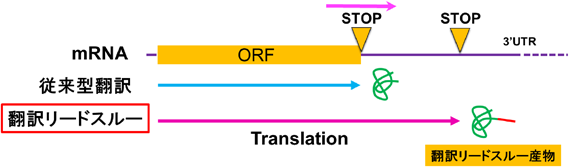
本稿では筆者らが新たに報告したL-MPZの性質など、これまでに明らかになってきたことについて紹介させていただく。
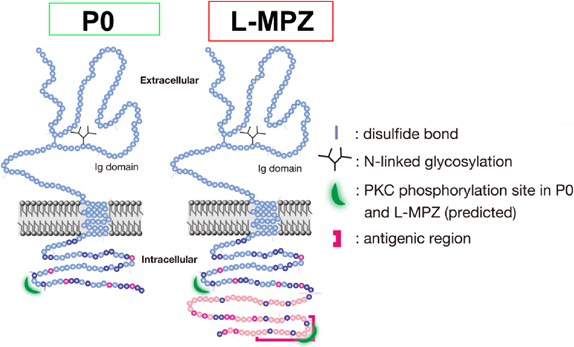
末梢神経髄鞘(ミエリン)は中枢神経系のミエリンとは異なり、シュワン細胞がミエリンを形成する。末梢神経ミエリンは脂質約70%とタンパク質約30%で構成される膜状構造物で軸索周囲を何重にも取り囲み、絶縁および軸索上イオンチャネルのランビエ絞輪部への局在化に関与し、跳躍伝導の発生に重要な役割を果たしている。最近では、ミエリンは跳躍伝導だけでなく神経系の発達過程や様々な神経活動に関与することが報告されている6)。末梢神経ミエリンタンパク質であるP0タンパク質は全末梢神経ミエリンタンパク質の約50%以上を占め、末梢神経ミエリンの形成や維持に非常に重要な約30 kDaの主要タンパク質である。P0タンパク質は細胞外にイムノグロブリン様ドメインを持つ1回膜貫通型の細胞接着糖タンパク質で、細胞内には接着性の調節に関わるProtein kinase C(PKC)リン酸化サイトを持っている(図2)。また、シャルコー・マリー・トゥース病1B型(CMT1B)に代表される重篤な遺伝性脱髄疾患の原因遺伝子としても知られている7–9)。CMTは原因遺伝子が特定されているものも多いが、原因分子も変異も多岐に渡り、発症メカニズムが不明なものがほとんどであり、根本的な治療法のない神経系難病に指定されている。
これまでにL-MPZは末梢神経障害を持つ患者の血清IgGに反応する正体不明の約36 kDaのP0関連タンパク質として報告されてきた10, 11)。しかし、P0タンパク質との明らかな分子量の違いなどがあり、この36 kDaのP0関連タンパク質についての詳細な構造や機能について明らかにされていなかった。そこで筆者らの研究室では末梢神経障害患者の血清に含まれる抗36 kDa IgG抗体を指標にして、この分子の解析を行った。
その結果、末梢神経ミエリン画分に濃縮されN-Link型糖鎖付加されているなど、P0タンパク質と同様の特徴を持つことが明らかとなった。次に特殊な2次元電気泳動法により分離したこの36 kDaのタンパク質バンドをトリプシン消化後にペプチドの質量分析を行ったが、P0タンパク質としてしか同定されなかった。しかし、これまでに36 kDa IgG抗体の抗原部位として3′非翻訳領域を含む可能性が示唆されていたことから11)、すでに報告されていたヒト、ウシ、ラット、マウスおよびカエルのP0 mRNAの終止コドン以降の配列に着目し比較してみた。その結果、P0本来の終止コドン(UAG)の次の終止コドン(UGA)までの塩基配列から予想される翻訳後の63アミノ酸の配列がヒトからカエルまで高い同一性を持ち系統学的に保存されていることが明らかとなった。その配列から予想されるトリプシン消化後ペプチドと同じ分子量のペプチドが前述の質量分析したデータに含まれていることも確認された。ヒトP0 cDNAを用いた強制発現系では、培養細胞においてもin vitro転写・翻訳系においても、同一のP0 cDNAからP0分子と36 kDa分子の両方が合成されることを明らかにした。また終止コドンのリードスルーを促進することが知られているアミノグリコシド系の試薬であるG418を使用すると36 kDa分子の合成が促進されることも明らかとなった。以上のことからP0 mRNAの本来の終止コドンのリードスルーにより次の終止コドンまで翻訳が行われ、C末側に63アミノ酸(分子量約6 kDa)が付加し36 kDa分子としてL-MPZが合成されていることが証明された3)。またC末側に新しく付加された63アミノ酸残基にはPKCによりリン酸化されるサイトがもう一つあることもわかり、これがL-MPZの機能に関連することが示唆された。
L-MPZはP0タンパク質と同一のmRNAより産生され、大部分がP0タンパク質と同様の構造を持つため、これまでに報告されていたP0タンパク質の機能の一部をL-MPZが担っている可能性がある。そこでP0タンパク質とL-MPZの機能を明らかにするために、L-MPZのみを発現するマウス(L-MPZマウス)の作製を行った。まず、生体内のP0タンパク質を全てL-MPZに置き換えるために、CRISPR-Cas9系のゲノム編集によりP0遺伝子の1つ目の本来のストップコドンTAGをAlanineのコドンであるGCGに変えたマウスを作製した。SDS-ポリアクリルアミドゲル電気泳動後のクマシー・ブリリアント・ブルー染色ならびに抗L-MPZ抗体と抗P0抗体によるウェスタンブロットでは、ヘテロ接合体では野生型に比べP0タンパク質の減少と共にL-MPZがほぼ等量まで増加していることが明らかとなった。一方、ホモ接合体ではP0タンパク質は欠損しL-MPZに全て置き換わっていることが確認できた(L-MPZ量、ホモ接合体(約100%)>ヘテロ接合体(約50%)>野生型(約5–10%))。次に、8~10週齡の成体マウスを用いて解析を行った。L-MPZマウスでは見かけ上の表現型に大きな変化がなかったため、運動機能の詳細を調べるために尻尾をつり下げたTail suspension test、Rotarod test、さらに電気生理学的神経伝導試験を行った。その結果、ホモ接合体マウスで、下肢を中心とした運動機能の低下、運動神経伝導速度や複合筋活動電位振幅の低下が認められた。また筋肉を観察してみると群萎縮や中心核などの神経原性の筋萎縮も認められた。
これらの結果から脱髄や軸索の異常が示唆されたため、次に坐骨神経の形態学的な観察を行った。その結果、異常な形態のミエリンや破壊されたミエリン、カハールバンドと呼ばれる物質輸送用として存在するシュワン細胞の細胞質を含む構造の異常、ランビエ絞輪周辺軸索上のイオンチャネル集積像の異常やランビエ絞輪間距離の短縮など多くの異常な所見が認められた。さらに末梢神経内で多くのマクロファージの浸潤も認められた。ウエスタンブロット解析では、小胞体ストレスマーカーの増加、ミエリン塩基性タンパク質(MBP)の低下が認められ、脱髄が示唆された。準超薄切片あるいは超薄切片の解析では、ミエリンを完全に消失した軸索や薄くなった有髄軸索の増加などのミエリンの異常だけではなく、大径有髄神経線維の減少(軸索の小径化)など軸索にも異常が認められた。さらに髄鞘において膜の密な重層化(コンパクション)の詳細を電子顕微鏡画像で解析した結果、脂質二重膜の細胞外側同士の接近により形成された周期間線:intraperiod line(IPL)は比較的保存されているのに対して、細胞膜内側同士の接点である周期線:major dense line(MDL)の開大が数多く見られた(図3上図)。他にも、L-MPZ量の増加に伴いPKCでリン酸化されたL-MPZが増加したことから、ミエリン形態の異常には、L-MPZの63アミノ酸の付加という物理的な大きさの増大とL-MPZ特異的ドメインのリン酸化による電気的な反発がMDLの開大を引き起した可能性が考えられた12)(図3下図)。一方で、ホモ接合体マウスに比べて明らかに軽度ではあるが、ヘテロ接合体マウスにおいても野生型に比べて運動機能や伝導速度の低下、異常な形態のミエリンが観察された。したがって、生体内でリードスルー調節に異常が生じ、通常よりもL-MPZ量が増加した場合には、ミエリンの異常による末梢神経障害を生じ得ることが明らかとなった12)。
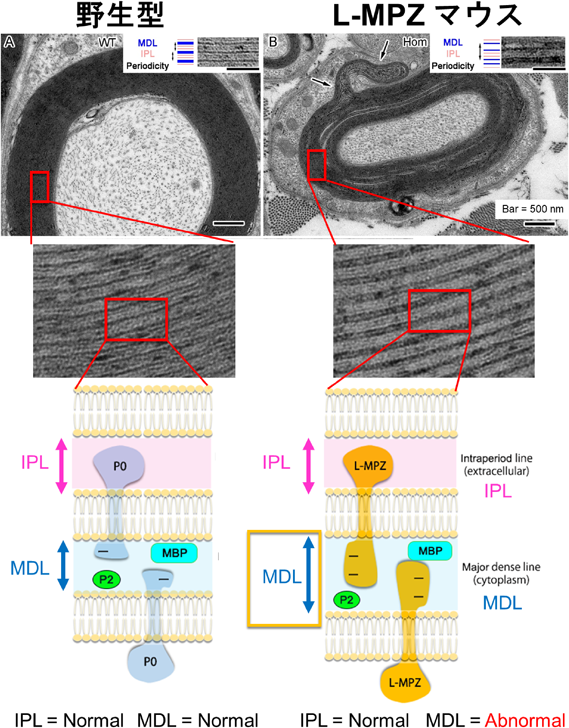
これまでにP0遺伝子はCMT1Bの原因遺伝子として知られており、この他にもDejerine-Sottas syndromeや軸索萎縮をともなうCMT2Iなどの遺伝病の一部の原因にもなっている。P0遺伝子を原因とする遺伝病には多様性があり、50に近い数の変異や欠損が報告されている8, 9)。その変異の多くは細胞接着に直接的に関与するN末側である細胞外ドメインに存在するが、C末側の膜貫通ドメインや細胞内ドメインの変異が原因でもCMTは発症する。これは細胞内ドメインにおける変異もPKCによるリン酸化の異常などを介し、細胞内輸送や細胞接着性に影響を及ぼすためだと考えられている13, 14)。これまでの研究により翻訳リードスルー産物であるL-MPZはP0タンパク質の細胞内のC末部分に63アミノ酸が付加した構造をしていること、L-MPZマウスの表現形がヒトのCMTで認められる末梢神経障害と一致することからL-MPZはいまだに原因が不明なCMTの病態に関与している可能性が示された。
現在、ヒトにおいてL-MPZが原因でCMTを発症するという報告はないが、L-MPZの配列が非翻訳領域にあるため解析が進んでいなかった可能性がある。今後、原因の明らかでないCMT患者のL-MPZ特異的配列の遺伝子の解析が必要だと考えられる。
本稿で紹介したL-MPZマウスはヒトのCMTのモデルとして有用であり、L-MPZマウスの発達過程や成体以降の病態の進行を詳細に調べることにより末梢神経障害の発症機序や進行性病態の解明につながると期待できる。また、L-MPZの異常や翻訳リードスルー調節の破綻によるL-MPZの異常産生などが、原因遺伝子の同定されていないCMTの発症に関与する可能性も考えられる。高等動物においてL-MPZ以外にも血管新生や中枢神経系に関わる分子の翻訳リードスルー産物が報告されていることから、翻訳リードスルーの破綻と病気との関連を調べることが重要になってくると考える。現在、L-MPZを産生せずP0タンパク質のみを発現するマウスの解析も行っており、L-MPZマウスとの比較解析によりL-MPZの生理的機能を解明できると期待している。
本稿で紹介した研究は昨年度で退職された東京薬科大学機能形態学教室の馬場広子先生および東京薬科大学機能形態学教室の山口宜秀先生をはじめとする、多くの先生方ならびに学生諸氏の御協力あってのことです。改めて御礼申し上げます。また、今回執筆の機会を与えてくださいました神経化学会出版・広報委員会、関係者の皆様に感謝致します。
1) Steneberg P, Englund C, Kronhamn J, Weaver TA, Samakovlis C. Translational readthrough in the hdc mRNA generates a novel branching inhibitor in the Drosophila trachea. Genes Dev, 12(7), 956–967 (1998).
2) Valle RPC, Drugeon G, Devignes-Morch MD, Legocki AB, Haenni AL. Codon context effect in virus translational readthrough A study in vitro of the determinants of TMV and Mo-MuLV amber suppression. FEBS Lett, 306(2-3), 133–139 (1992).
3) Yamaguchi Y, Hayashi A, Campagnoni CW, Kimura A, Inuzuka T, Baba H. L-MPZ, a Novel Isoform of Myelin P0, Is Produced by Stop Codon Readthrough. J Biol Chem, 287(21), 17765–17776 (2012).
4) Eswarappa SM, Potdar AA, Koch WJ, Fan Y, Vasu K, Lindner D, Willard B, Graham LM, DiCorleto PE, Fox PL. Programmed Translational Readthrough Generates Antiangiogenic VEGF-Ax. Cell, 157(7), 1605–1618 (2014).
5) De Bellis M, Pisani F, Mola MG, Rosito S, Simone L, Buccoliero C, Trojano M, Nicchia GP, Svelto M, Frigeri A. Translational readthrough generates new astrocyte AQP4 isoforms that modulate supramolecular clustering, glial endfeet localization, and water transport. Glia, 65(5), 790–803 (2017).
6) Nave KA. Myelination and the trophic support of long axons. Nat Rev Neurosci, 11(4), 275–283 (2010).
7) Hayasaka K, Himoro M, Sato W, Takada G, Uyemura K, Shimizu N, Bird TD, Conneally PM, Chance PF. Charcot-Marie-Tooth neuropathy type 1B is associated with mutations of the myelin P0 gene. Nat Genet, 5(1), 31–34 (1993).
8) Shy ME. Peripheral neuropathies caused by mutations in the myelin protein zero. J Neurol Sci, 242(1-2), 55–66 (2006).
9) Sanmaneechai O, Feely S, Scherer SS, Herrmann DN, Burns J, Muntoni F, Li J, Siskind CE, Day JW, Laura M, Sumner CJ, Lloyd TE, Ramchandren S, Shy RR, Grider T, Bacon C, Finkel RS, Yum SW, Moroni I, Piscosquito G, Pareyson D, Reilly MM, Shy ME; Inherited Neuropathies Consortium - Rare Disease Clinical Research Consortium (INC-RDCRC). Genotype-phenotype characteristics and baseline natural history of heritable neuropathies caused by mutations in the MPZ gene. Brain, 138(Pt 11), 3180–3192 (2015).
10) Meléndez-Vásquez CV, Gregson NA. Characterization and partial purification of a novel 36 kDa peripheral myelin protein recognized by the sera of patients with neurological disorders. J Neuroimmunol, 91(1-2), 10–18 (1998).
11) Ishida K, Takeuchi H, Takahashi R, Yoshimura K, Yamada M, Mizusawa H. A possible novel isoform of peripheral myelin P0 protein: a target antigen recognized by an autoantibody in a patient with malignant lymphoma and peripheral neuropathy. J Neurol Sci, 188(1-2), 43–49 (2001).
12) Otani Y, Ohno N, Cui J, Yamaguchi Y, Baba H. Upregulation of large myelin protein zero leads to Charcot—Marie—Tooth disease-like neuropathy in mice. Commun Biol, 3(1), 121 (2020).
13) Xu W, Shy M, Kamholz J, Elferink L, Xu G, Lilien J, Balsamo J. Mutations in the cytoplasmic domain of P0 reveal a role for PKC-mediated phosphorylation in adhesion and myelination. J Cell Biol, 155(3), 439–446 (2001).
14) Konde V, Eichberg J. Myelin protein zero: Mutations in the cytoplasmic domain interfere with its cellular trafficking. J Neurosci Res, 83(6), 957–964 (2006).

This page was created on 2021-06-08T17:02:29.624+09:00
This page was last modified on 2021-07-02T15:33:32.000+09:00
このサイトは(株)国際文献社によって運用されています。