Racシグナルの破綻による神経発達障害発症機構の解明
名古屋大学 細胞制御学グループ
発行日:2023年12月30日Published: December 30, 2023
© 2023 日本神経化学会© 2023 The Japanese Society for Neurochemistry
神経発達障害は、認知・学習・社会性等の能力に偏りや問題を生じ、現実生活に困難をきたす障害である。その原因の一つとして、中枢神経系における神経細胞の適切な発生/増殖→移動による皮質層構造形成→軸索/樹状突起分化によるシナプスネットワーク構築プロセスの破綻が挙げられる。遺伝子異常による内的要因と、母体の妊娠/出生時の感染症・低酸素・血液循環障害などによって起きる外的要因が知られるが、近年のシーケンス技術発展により前者の病態メカニズム研究が盛んに行われている。
Racは、細胞内シグナル伝達と細胞骨格ダイナミクスを制御するRhoファミリー低分子量Gタンパク質の一角である1, 2)。全ての真核生物において保存されているRhoファミリーは、細胞周期・接着・遊走などの基本的な細胞イベントにおいて必須で、神経発生/発達過程にも関与することが知られている2)。Racは、典型的なGタンパク質と同様に、細胞内では活性型/GTP(グアニンヌクレオチド-3-リン酸)結合型と、不活性化型/GDP(グアニンヌクレオチド-2-リン酸)結合型を循環する分子スイッチとして働いている。このGサイクルは、主にグアニンヌクレオチド交換因子(GEF)とGTPase活性化タンパク質(GAP)により厳密に制御されている。GEFは、Gタンパク質からのGDP解離を触媒し、GTP結合型への移行を促進するのに対して、GAPは、Gタンパク質の内在性GTPase活性を賦活化ことで、GDP結合型への移行を促す3)。GTP結合型Racは、様々な下流エフェクターと選択的に相互作用することで、アクチン細胞骨格ダイナミクス、遺伝子転写活性制御などを担う。近年、知的障害患者からヒトRACとその関連分子群の遺伝子バリアントが複数同定され、Racシグナルと神経疾患の連関が着目されている。
Racサブファミリーは、相同性約90%のRac1-3からなる。これらの発現分布は、Rac1がユビキタスに発現しているのに対して4)、Rac2は造血細胞系に特異的に発現しており5)、Rac3は発生/発達中および成体の中枢神経系に多く発現している6, 7)。近年、知的障害患者から、ヒトRAC1およびRAC3のde-novoバリアントが複数同定された8–11)。RAC1/3バリアントは、発生/発達時の神経細胞のアクチン細胞骨格再編成を伴う発生/分化(移動・突起伸長/分岐形成)に直接的な有害作用をもたらすと想定される。また、その特異的GEFであるTRIO, PLEKHG2、下流エフェクターであるPAKでも遺伝子バリアントが複数例見つかり、神経発達障害との関連が示唆された12–14)。筆者ら研究グループはこれらの臨床例から、RACの調節異常やエフェクター異常によるシグナル破綻が神経発達障害発症に関わるという新たな神経疾患概念“Neuro-RACopathy”を提唱した15)。ただし、臨床症状は一様ではなく各種遺伝子・バリアント依存的に異なるため、発症メカニズムを解明するためには、各タンパク質の神経生理機能と、バリアント性状(Gain-of-function, Loss-of-function, Dominant negativeタイプか?)を検証・解析しなければならない。
本稿では、小頭症患者から同定されたPLEKHG2バリアントと、皮質形成異常症患者から同定されたRAC3バリアントの研究例を紹介する。当時、RAC3とPLEKHG2の詳細な神経生理機能はわかっておらず、さらに同じRacシグナル異常にも関わらず臨床症状に劇的な違いが生じる理由も全く不明であった。そこで、PLEKHG2異常症とRAC3異常症の病態メカニズムの“違い”を明らかにすることで、Racシグナル破綻による発達障害の発症機序を解明することを目的に研究を実施した。
2016年に、重度精神遅滞・出生後小頭症を有する患者からPLEKHG2 p.R204Wのホモ接合性バリアントが同定された13)。PLEKHG2はDblファミリーRac-GEFとして知られており、Rac活性化を介してアクチン細胞骨格制御、転写制御などを担う分子として知られる16)。R204は、PLEKHG2の酵素活性中心(Dbl homologyドメイン102-283)に属している。また、PLEKHG2-Rac1複合体の予想構造(PDB: 5FI0ベースのSWISS-MODEL)は、R204がRac1結合界面にあることを示した。そのため、当バリアントはPLEKHG2のRac活性化機能に大きな影響を与えることが予想された。
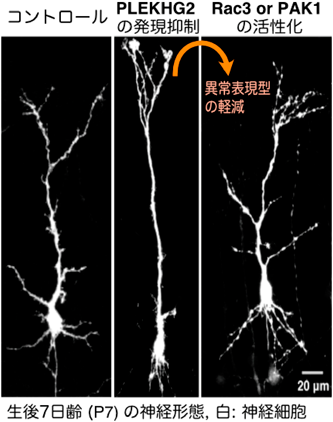
筆者らが、p.R204Wバリアントの生化学的性状を検討した結果、PLEKHG2 p.R204WのRac1, Rac3に対するグアニンヌクレオチド交換活性が、野生型に比べて低下していることがわかった。さらに、Rac1, Rac3を介したPAK1への活性化シグナル(PLEKHG2→Rac1/3→PAK1)も低下していたため、p.R204WはLoss-of-Function型変異であることがわかった。PLEKHG2はマウス胎仔~成獣の大脳皮質神経細胞で発現しており、初代培養神経細胞では細胞質・軸索・樹状突起・樹状突起上スパインに局在することが示されている17)。このことから、PLEKHG2が神経発生/発達に寄与することが想定され、Loss-of-Functionの影響を検証する必要があった。そこで、マウス子宮内胎仔脳電気穿孔法(IUE)を用い、マウスPLEKHG2ホモログの発現を低下させた病態モデルを作成した。胎生期14日目でPLEKHG2をノックダウンしたとき、神経幹細胞発生/増殖と、神経細胞移動ではコントロールとの相違は観られなかった。一方で、7日齢マウスの神経細胞において顕著な樹状突起形成不全が観察され、14日齢マウスの神経細胞では、樹状突起上のスパイン密度が低下した(図1)。さらに、PLEKHG2発現低下による神経細胞の樹状突起形成障害は、下流シグナル分子であるRac3、もしくはPAK1を発現させると改善されることが示された(図1)。また、PLEKHG2ノックダウンは7日齢時点のマウス神経細胞の脳梁軸索投射を阻害するが、初代培養マウス皮質神経細胞でPLEKHG2をノックダウンしても軸索突起伸長が阻害された。このことから、PLEKHG2による神経突起の形態制御はcell-autonomousな現象であることがわかった。以上の結果から、神経細胞の樹状突起・軸索分化形成過程においてPLEKHG2→Rac→PAK1シグナルが重要であり、変異によるシグナル不全/制御破綻が神経分化障害を引き起こすことが示唆された18)(図3)。
ユビキタスな発現プロファイルを示すRac1と比較して、Rac3は中枢神経系に限局して発現している。筆者らは、Rac3がマウス大脳皮質、および海馬興奮性ニューロンの軸索・樹状突起・樹状突起上スパインに発生時間依存的に局在することを観察した19)。このことから、Rac3は神経発生/発達において重要な役割を担うことが強く示唆された。既報のRAC3バリアントp.P29L, p.P34R, p.Q61L, p.E62Kに加えて10, 11)、皮質形成異常症を伴う知的障害患者から新規変異p.G12R, p.F28S, p.A59G, p.G60D, p.E62del, p.D63N, p.Y64C, p.K116Nが、欧米を中心とする国際他機関共同研究グループによって同定された。Gタンパク質-ヌクレオチド結合とGTP水解活性は、ファミリー間で高度に保存されたGドメイン(G1–5)によって仲介される。特に、G2とG3に跨るSwitch IとSwitch IIという2つの領域は、GDP/GTP結合依存的に“off/on”の分子構造変化を起こす。GTP結合状態では、様々な下流エフェクターと選択的に相互作用するためのプラットフォームを提供し、細胞内シグナルカスケードを開始することができる。特にRacは、アクチン細胞骨格の分岐形成シグナルを促進し、葉状仮足(膜ラッフリング)形成に寄与することで有名である。これらのドメインは、上記ヒトRAC3バリアントのホットスポットになっている。
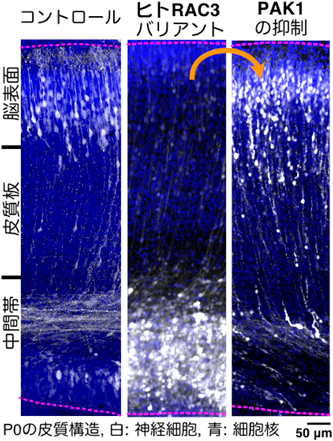
そこで筆者らは、12種類のRAC3バリアントがGタンパク質活性に影響を与えると予想し、生化学的性状(GDP/GTP交換速度、GTP水解活性、エフェクター結合アッセイ)を評価した。その結果、12種のバリアントはすべてGain-of-Function型であることを明らかにした。次に、初代培養神経細胞に各種バリアントを発現させると、顕著な葉状仮足形成と極性消失が観察された。さらに、細胞内シグナル解析では、バリアントに依存して種々の下流シグナル経路(MAP、NF-kB、アクチン関連シグナル)を複合的に障害すること、PAK1シグナルは顕著に増強されることを示した。次に、IUEを用いて、各種RAC3バリアントを発現させたマウス病態モデルを作成し、in vivoにおける神経発生/発達への影響を観察した。胎生14日目に遺伝子導入し、胎生16日目で神経細胞のスライス培養ライブイメージングをした結果、脳室帯→中間体→大脳皮質板移動時の細胞形態変化(多極性→双極性への移行)に障害が生じ、顕著な神経細胞移動障害が観察された。また、0日齢、7日齢時点でも神経細胞の異所性局在が観察され、皮質層構造の破綻がみられた(図2)。一方、RAC3バリアント発現細胞の移動障害はPAK1を阻害すると改善されることも示した(図2)。すなわち、皮質層形成時の神経細胞移動過程において、Rac3→PAK1シグナルが重要であり、バリアントによるシグナル過剰活性化/制御破綻が神経細胞の形態・極性崩壊を引き起こすことが示唆された20, 21)(図3)。
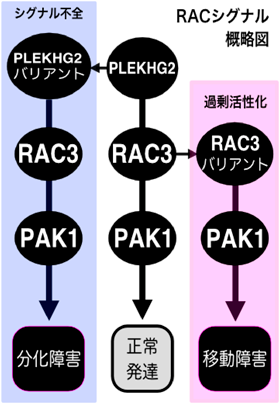
RhoファミリーG蛋白質の代表格であるRacであるが、その知見の主たるはin vitroでのアクチン骨格の制御因子としての細胞生物学的機能や癌との関連についてであり、in vivo(哺乳類)における神経機能に言及する報告は意外と少ない。また、知的障害発症機序はシナプスネットワーク構造の破綻が病因だと短絡的に言及される一方で、申請者は神経細胞発達過程の何れの破綻かまで追求しなければ核心に迫ることができないと考えた。その結果、PLEKHG2異常症(小頭症)とRAC3異常症(皮質形成異常症)の臨床症状の違いに着目し、Racシグナル不全では神経移動正常/分化異常、シグナル過剰では神経移動異常による皮質形成障害、というRacの絶妙な制御バランスが神経発達に重要という興味深い見解に辿り着いた。さらに、エフェクター分子のPAK1の活性調節にてPLEKHG2、RAC3病態を共に軽減できることも示した。すなわち、PAK1は治療シーズになると期待され、これら知見は大きな臨床的意義がある。
今後は、小頭症・巨脳症の責任分子Rac1(バリアント特異的に脳サイズが変化する興味深い分子)や、自閉症責任分子PREX1(Rac-GEF)などを研究対象にし、神経発生/発達におけるRac機能を俯瞰的に検証したいと考えている。これら成果により、“Neuro-RACopathy”の病因エフェクター(マスター分子)を同定することができれば、小児発達障害患者に対する治療薬開発の礎になると期待される。
本稿で紹介した研究成果は、愛知県医療療育総合センター発達障害研究所で得られました。多大なるご指導を賜りました永田浩一部長、伊東秀記室長、田畑秀典室長をはじめ研究所の皆様に厚く御礼申し上げます。また、筆者を研究者として育てていただいた岐阜大学 上田浩教授、これまでご指導いただきました諸先生方、本稿の執筆機会を与えて下さいました日本神経化学会の先生方、および関係者の皆様に深く感謝いたします。今後も奨励賞受賞者としての自覚を持ち、名古屋大学 木下専教授のもとで神経化学研究に精進してまいります。
1) Hall A. Rho GTPases and the actin cytoskeleton. Science, 279(5350), 509–514 (1998).
2) Stankiewicz TR, Linseman DA. Rho family GTPases: Key players in neuronal development, neuronal survival, and neurodegeneration. Front Cell Neurosci, 8, 1–14 (2014).
3) Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol, 6(2), 167–180 (2005).
4) Moll J, Sansig G, Fattori E, van der Putten H. The murine rac1 gene: cDNA cloning, tissue distribution and regulated expression of rac1 mRNA by disassembly of actin microfilaments. Oncogene, 6(5), 863–866 (1991).
5) Shirsat NV, Pignolo RJ, Kreider BL, Rovera G. A member of the ras gene superfamily is expressed specifically in T, B and myeloid hemopoietic cells. Oncogene, 5(5), 769–772 (1990).
6) Malosio ML, Gilardelli D, Paris S, Albertinazzi C, De Curtis I. Differential expression of distinct members of rho family GTP-binding proteins during neuronal development: Identification of RAC1B, a new neural- specific member of the family. J Neurosci, 17(17), 6717–6728 (1997).
7) Haataja L, Groffen J, Heisterkamp N. Characterization of RAC3, a novel member of the Rho family. J Biol Chem, 272(33), 20384–20388 (1997).
8) Margot RF, Ansor NM, Kousi M, Yue WW, Tan PL, Clarkson K, Clayton-Smith J, Corning K, Jones JR, Lam WWK, Mancini GMS, Marcelis C, Mohammed S, Pfundt R, Roifman M, Cohn R, Chitayat D, Millard TH, Katsanis N, Brunner HG, Banka S. RAC1 Missense Mutations in Developmental Disorders with Diverse Phenotypes. Am J Hum Genet, 101(3), 466–477 (2017).
9) Banka S, Bennington A, Baker MJ, Rijckmans E, Clemente GD, Ansor NM, Sito H, Prasad P, Anyane-Yeboa K, Badalato L, Dimitrov B, Fitzpatrick D, Hurst ACE, Jansen AC, Kelly MA, Krantz I, Rieubland C, Ross M, Rudy NL, Sanz J, Stouffs K, Xu ZL, Malliri A, Kazanietz MG, Millard TH. Activating RAC1 variants in the switch II region cause a developmental syndrome and alter neuronal morphology. Brain, 145(12), 4232–4245 (2022).
10) Hiraide T, Kaba Yasui H, Kato M, Nakashima M, Saitsu H. A de novo variant in RAC3 causes severe global developmental delay and a middle interhemispheric variant of holoprosencephaly. J Hum Genet, 64(11), 1127–1132 (2019).
11) Costain G, Callewaert B, Gabriel H, Tan TY, Walker S, Christodoulou J, Lazar T, Menten B, Orkin J, Sadedin S, Snell M, Vanlander A, Vergult S, White SM, Scherer SW, Hayeems RZ, Blaser S, Wodak SJ, Chitayat D, Marshall CR, Meyn MS. De novo missense variants in RAC3 cause a novel neurodevelopmental syndrome. Genet Med, 21(4), 1021–1026 (2019).
12) Pengelly RJ, Greville-Heygate S, Schmidt S, Seaby EG, Jabalameli MR, Mehta SG, Parker MJ, Goudie D, Fagotto-Kaufmann C, Mercer C, Debant A, Ennis S, Baralle D; DDD Study. Mutations specific to the Rac-GEF domain of TRIO cause intellectual disability and microcephaly. J Med Genet, 53(11), 735–742 (2016).
13) Edvardson S, Wang H, Dor T, Atawneh O, Yaacov B, Gartner J, Cinnamon Y, Chen S, Elpeleg O. Microcephaly-dystonia due to mutated PLEKHG2 with impaired actin polymerization. Neurogenetics, 17(1), 25–30 (2016).
14) Harms FL, Kloth K, Bley A, Denecke J, Santer R, Lessel D, Hempel M, Kutsche K. Activating Mutations in PAK1, Encoding p. 21-Activated Kinase 1, Cause a Neurodevelopmental Disorder. Am J Hum Genet, 103(4), 579–591 (2018).
15) Scala M, Nishikawa M, Nagata K, Striano P. Pathophysiological Mechanisms in Neurodevelopmental Disorders Caused by Rac GTPases Dysregulation: What’s behind Neuro-RACopathies. Cells, 10(12), 3395 (2021).
16) Ueda H, Nagae R, Kozawa M, Morishita R, Kimura S, Nagase T, Ohara O, Yoshida S, Asano T. Heterotrimeric G protein betagamma subunits stimulate FLJ00018, a guanine nucleotide exchange factor for Rac1 and Cdc42. J Biol Chem, 283(4), 1946–1953 (2008).
17) Nishikawa M, Ito H, Noda M, Hamada N, Tabata H, Nagata K. Expression analyses of PLEKHG2, a Rho family-specific guanine nucleotide exchange factor, during mouse brain development. Med Mol Morphol, 54(2), 146–155 (2021).
18) Nishikawa M, Ito H, Tabata H, Ueda H, Nagata K. Impaired Function of PLEKHG2, a Rho-Guanine Nucleotide-Exchange Factor, Disrupts Corticogenesis in Neurodevelopmental Phenotypes. Cells, 11(4), 696 (2022).
19) Nishikawa M, Ito H, Noda M, Hamada N, Tabata H, Nagata K. Expression analyses of Rac3, a Rho family small GTPase, during mouse brain development. Dev Neurosci, 44(1), 49–58 (2022).
20) Nishikawa M, Scala M, Umair M, Ito H, Waqas A, Striano P, Zara F, Costain G, Capra V, Nagata K. Gain-of-function p.F28S variant in RAC3 disrupts neuronal differentiation, migration and axonogenesis during cortical development, leading to neurodevelopmental disorder. J Med Genet, 60(3), 223–232 (2023).
21) Scala M, Nishikawa M, Ito H, Tabata H, Khan T, Accogli A, Davids L, Ruiz A, Chiurazzi P, Cericola G, Schulte B, Monaghan KG, Begtrup A, Torella A, Pinelli M, Denommé-Pichon AS, Vitobello A, Racine C, Mancardi MM, Kiss C, Guerin A, Wu W, Gabau Vila E, Mak BC, Martinez-Agosto JA, Gorin MB, Duz B, Bayram Y, Carvalho CMB, Vengoechea JE, Chitayat D, Tan TY, Callewaert B, Kruse B, Bird LM, Faivre L, Zollino M, Biskup S, Brown G, Butte MJ, Dell’Angelica EC, Dorrani N, Douine ED, Fogel BL, Gutierrez I, Huang A, Krakow D, Lee H, Loo SK, Mak BC, Martin MG, Martínez-Agosto JA, McGee E, Nelson SF, Nieves-Rodriguez S, Palmer CGS, Papp JC, Parker NH, Renteria G, Sinsheimer JS, Wan J, Wang L, Perry KW, Nigro V, Brunetti-Pierri N, Casari G, Cappuccio G, Torella A, Pinelli M, Musacchia F, Mutarelli M, Carrella D, Vitiello G, Capra V, Parenti G, Leuzzi V, Selicorni A, Maitz S, Banfi S, Zollino M, Montomoli M, Milani D, Romano C, Tummolo A, De Brasi D, Coppola A, Santoro C, Peron A, Pantaleoni C, Castello R, D’Arrigo S, Striano P, Nigro V, Severino M, Capra V, Costain G, Nagata K. Variant-specific changes in RAC3 function disrupt corticogenesis in neurodevelopmental phenotypes. Brain, 145(9), 3308–3327 (2022).

This page was created on 2023-11-07T09:37:52.111+09:00
This page was last modified on 2024-01-05T08:54:28.000+09:00
このサイトは(株)国際文献社によって運用されています。