リソソーム膜破綻とリソファジー機能低下が異常α-シヌクレインの伝播を引き起こす
大阪大学大学院医学系研究科 神経内科学教室
発行日:2024年6月30日Published: June 30, 2024
© 2024 日本神経化学会© 2024 The Japanese Society for Neurochemistry
神経変性疾患は中枢神経系の特定の神経細胞群が健常人よりも早期かつ急速に減少する疾患の総称であり、人口の高齢化に伴い世界的に急増している。多くは孤発性で遺伝的背景と環境因子により引き起こされる多因子疾患であり根本的な原因は未解明であるが、これまでの遺伝学研究および病理学研究によりアルツハイマー病のアミロイドβ、パーキンソン病(PD)のαシヌクレイン(αSyn)、筋萎縮性側索硬化症(ALS)のTDP43など疾患毎にそれぞれ特定のタンパク質が異常構造をとり凝集体を形成して伝播・蓄積することが病態の中心と考えられるようになってきた。本稿ではPDを中心に、どのようにして異常タンパクが中枢神経系を伝播・蓄積するかについて筆者らの研究成果を交えて概説する。
PDは国内でおよそ15–20万人が罹患するアルツハイマー病についで多い神経難病である。中脳黒質のドパミン神経を主体に神経細胞が脱落し、振戦(ふるえ)、運動緩慢などの運動症状と自律神経症状や認知機能障害などの非運動症状を進行性に呈する。神経変性は不可逆で現在までにその進行を回復することはおろか、停止ないし緩和する手段(疾患修飾治療)は得られておらず治療は対症療法に限られる。病理学的には神経細胞質に蓄積するLewy小体により定義され、1997年にLewy小体の主要構成成分がαSynであること1)とαSynをコードするSNCA遺伝子が家族性PD(PARK1)の原因遺伝子であることが発見された2)。ここからαSynの異常がPDの原因であると考えられて研究が発展し、病理学的にはPDの病期に対応して脳内のLewy病理が嗅球あるいは延髄から始まり中脳および大脳辺縁系そして大脳皮質に拡大すること(Braak仮説)が示され3)、生化学的には異常なβシート構造となり凝集したαSynが正常αSynを異常構造に変換し更なる凝集体をつくること(プリオン仮説)が示された4)。これらの結果から今日では脳内で異常αSynが神経細胞間を伝わり正常のαSynを巻き込みながら伝播することがPDの原因として有力視されている。これまでにαSyn単量体そのものをターゲットとして抗体医薬等の疾患修飾治療の開発が試みられてきたが未だ実現には至っていない。我々はαSyn病態のさらなる理解を目指して、αSynの細胞間での伝播の機序とそれに対する防御機構について研究を進めてきた。
リソソームは細胞内の有害物の分解の場であり、細胞外からとりこまれた有害物はエンドサイトーシスによりリソソームへ輸送され、細胞質の損傷した細胞小器官や蓄積したタンパク凝集体はオートファジーによりリソソームへ輸送される。実験的にはマクロオートファジーおよびシャペロン介在性オートファジーがαSynの分解経路であり動物モデルでオートファジーの機能低下が異常αSynの蓄積と運動機能低下を引き起こすことが示されている5)。遺伝学的にはリソソーム酵素の一つで糖脂質の分解を担うグルコセレブロシダーゼをコードするGBA1遺伝子を始め、複数のリソソーム関連の遺伝子がPDの遺伝的リスクとして同定された6)。剖検脳の検討ではPD患者においてリソソーム機能が低下しており、Lewy病理の大部分がオートファジー関連タンパク質LC3を含むことが報告されている7)。これらの結果からオートファジー–リソソーム系の機能低下が異常αSynの蓄積を誘導すると考えられるが、逆に異常αSynの増加がオートファジーフラックスやCMAを妨げることも報告されている8)。筆者らはオートファジー研究の第一人者である大阪大学吉森教授らとの共同研究によりPD病態とオートファジー機能低下について研究を進めてきた。その中でオートファジーを負に制御するRubiconが加齢に伴って蓄積しており、このRubiconを抑制するとオートファジー機能が改善し線虫モデルおよびマウスモデルにおいて異常αSynの蓄積が軽減することが明らかとなった9)。またオートファジーの分解過程で生じる中間体アンフィソームとリソソームの融合を制御する因子PACSIN1の機能低下により異常αSynの蓄積が増強することも確認された10)。これらから加齢に伴うオートファジー–リソソーム系の機能障害が神経疾患における異常タンパクの蓄積に重要であると考えられる。
前述したように異常αSynは神経細胞間を伝播すると考えられているが、その仕組みについてはまだ不明な点が多い。これまでに細胞外のαSyn凝集体はいくつかの受容体を介してエンドサイトーシスにより神経細胞に取り込まれることが報告されているが11)、エンドサイトーシスで取り込まれたαSyn凝集体はリソソームに至り、細胞質に発現する正常αSynからはリソソーム膜で隔てられている。このためどのようにして取り込まれた凝集体がリソソーム内から細胞質のαSynへ凝集伝播するかは不明であった。近年、αSynを含めた神経難病の原因となる蛋白凝集体は小胞膜を破る性質があることが報告され12)、リソソーム膜の破綻による凝集体の漏出が伝播経路として有力視されていた。
我々はリソソーム膜破綻と凝集の伝播の関連について明らかにすべく、実験的に人工的に作成したαSyn凝集体(pre-formed fibril: PFF)を投与した培養細胞で伝播により細胞内の正常αSynが凝集する過程を可視化し、その過程におけるリソソーム膜破綻の意義を検討した。その結果、PFFは投与24時間後にはリソソームに集積して、一部のリソソームで膜損傷を起こし、一細胞質のαSyn凝集を反映するリン酸化αSyn(pSyn)が陽性となった。pSynは膜損傷のマーカーGal3、リソソームマーカーLAMP1が共局在を示し、さらに細胞質のαSyn凝集過程を過剰発現させたαSyn-EGFPにより可視可するとαSyn-EGFPの集積はPFFを含むリソソーム近傍に始まることが分かった。これらの結果からαSyn凝集体はエンドサイトーシスされリソソームへ運搬された後に、一部でリソソームの膜損傷を起こして細胞質へ漏出し凝集を伝播することが示された。更に人為的にリソソーム膜破綻を高率に起こす試薬(LLOMe)を用いて損傷リソソームを増加させるとPFF投与による細胞質のpSyn=凝集の伝播が有意に増加することからもこの説が支持された13)(図1)。
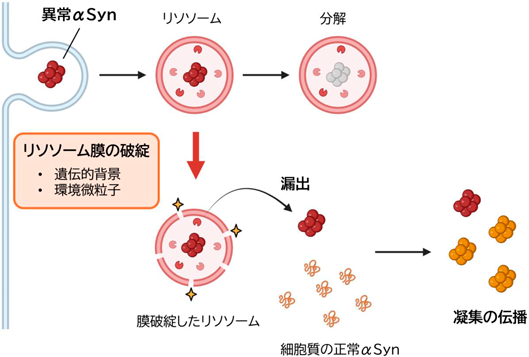
実験的にはαSyn凝集体によるリソソームの損傷は緩徐で少数であったため、異常αSyn自体によるリソソーム膜の損傷以外にPD病態において他の要因によりリソソーム膜脆弱性が加わっていることも想定された。
まずPDの遺伝的因子としてGBA1変異に着目した。前述したようにGBA1変異がPDの最大の遺伝学的リスクであるが、その病態機序としてはグルコセレブロシダーゼの機能低下に伴い蓄積するグルコシルセラミドとαSynの相互作用、異常グルコセレブロシダーゼの蓄積による小胞体ストレス、ミトコンドリア機能異常などが考えられてきた。またGBA1変異は他のリソソーム酵素の活性の低下やpHの上昇などの広範なリソソーム機能異常を来すことが報告されており14)、我々は先の結果で得られたリソソーム膜の脆弱性に着目し検証を行った。するとGBA1をノックダウンした細胞では定常状態およびPFFの投与下でいずれも野生型細胞よりも多く膜損傷を示すGal3陽性細胞が見られ、PFF投与時のLC3の集積(リソファジー)も多く、さらに凝集の伝播を示すpSyn陽性細胞も増加した。これらの結果からGBA1変異による病態においてリソソーム膜の脆弱性による凝集伝播の増強も寄与していることが示された。
一方で疫学研究から大気汚染や農薬などの環境因子がPDの発症に関わることがこれまでに知られていた。特に大気中のPM2.5の濃度とPDの発症率に相関が見られることが報告されており近年注目を集めている15)。我々は金沢大学瀬戸研究室で大気中から採取されたPM2.5の供与を受けて、実際のPM2.5がリソソーム膜傷害能を有すること、PM2.5の成分の中ではシリカが特に傷害性が高いことを確認した。更にPFFと併せてシリカを投与すると広範なリソソーム損傷に伴って細胞質でのαSyn凝集が促進された。これらの結果からGBA1変異やPM2.5等のPDの遺伝的背景および環境因子はリソソーム膜の脆弱性を惹起して異常αSynの伝播に寄与することが示された。
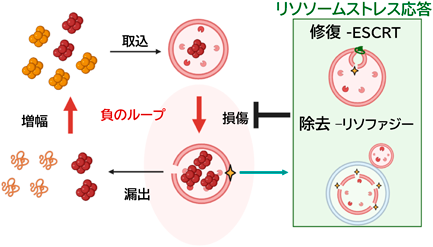
このようにリソソーム膜の脆弱性がPD病態において重要であることが示唆されたが、損傷を受けたリソソームからの高濃度のプロトンや分解酵素の細胞質への漏出は有害であり、リソソームが損傷を受けた際に働く複数の防御機構がリソソームストレス応答として報告されている。具体的には1. リソソーム修復、2. 選択的オートファジーによる隔離・除去(リソファジー)、3. リソソーム新生からなる16)。リソソーム修復では主にESCRT(Endosomal Sorting Complex Required for Transport)によるものの解明が進んでいる。ESCRTは小胞輸送における膜の切り離しを担うタンパク複合体として知られていたが、リソソーム損傷後すぐ(数分以内)にCaの流出を検知してESCRT-0、-I、-II、Alixなどの分子が損傷部位に誘導され、最終的にESCRT-IIIにより損傷部位を含む膜が内腔側に嵌入してくびりきられる17)。修復は主に小さな損傷に対して働き、大きな損傷をうけたリソソームは選択的オートファジー=リソファジーによりオートファゴソームによって細胞質から隔離され、オートファゴソームと別のリソソームが融合することにより隔離・除去される18)。これらの分子機構の多くは共同研究者である大阪大学吉森教授らのグループから報告されており、我々はこれらのリソソーム損傷応答がリソソーム膜破綻を介するαSyn凝集体の伝播において防御的な役割をもつのではないかとの仮説の下に研究を進めた。
その結果、PFFを投与した細胞ではリソソーム上にLC3の集積が起こり、電子顕微鏡観察と併せて実際にリソファジーが起きていることを確認した。次にリソファジーの防御的役割を検証するためにオートファジーレギュレーターのFIP200遺伝子をノックアウトした細胞(FIP200KO)にPFF処理をすると、正常細胞よりも有意にGal3およびpSyn陽性細胞が増加する事を確認した。すなわち細胞外から取り込まれた異常αSynがリソソーム膜損傷を起こして細胞質へ漏出し凝集が伝播することを、リソファジーが損傷リソソームを隔離・除去することによって防御していることが示唆された。また次にESCRTによるリソソーム損傷についても検証した。PFF投与24時間後ではESCRT-IIIの一種CHMP4Bのリソソーム上の集積が有意ではないが増加する傾向にあり、異常αSynによる緩徐なリソソーム損傷に対しての修復応答を反映していると考えられた。野生型細胞においてAlixおよびTSG101のノックダウンによりリソソーム修復を抑制するとαSyn凝集の伝播が増悪することを確認した。一方でリソファジーが働かないFIP200KOではAlix/TSG101のノックダウンで差が見られず、リソソーム修復もαSyn凝集の伝播に防御的に働くものの、併せてリソファジーが機能することが必要であることが示唆された。さらにLLOMeによるリソソーム膜の破綻とリソファジーの機能低下(FIP200KO)を同時に行うと、相乗的にαSyn凝集の伝播が促進されることも確認し、マウス初代神経細胞を用いたαSyn凝集の伝播モデルでも同様の結果が得られた。これまでの結果から、異常αSyn凝集の伝播はリソソーム膜の破綻により異常αSynが漏れ出すことを起点としておりリソソームストレス応答が損傷リソソームを修復あるいは除去することにより防御的に働いていることが示された。また海外からは別の神経変性疾患であるアルツハイマー病におけるタウ蛋白の蓄積においても同様にリソソームストレス応答が防御的に働くことが報告されている19, 20)。したがってリソソームストレス応答は異常タンパクの蓄積を伴う神経変性疾患に対して広く有効な治療標的となることが示唆される。
中枢神経細胞は生涯にわたり分裂・増殖しないためオートファジー・リソソーム系による細胞内の恒常性維持が極めて重要である。PDを含めて神経変性疾患は加齢に伴い発症率が劇的に増加し、ここには老化による環境中の有害物の蓄積によるリソソーム傷害やリソソームストレス応答の機能低下が関与していると考えられる。筆者らの研究によりリソソーム損傷と異常タンパクの蓄積が負のループを形成して病態を促進することが示唆され、リソソームストレス応答によるリソソーム恒常性の回復が治療の鍵となる。神経変性疾患の克服に向けて病態におけるリソソームストレス応答の更なる機序解明とその活性化による治療開発を目指したい。
本研究を行うにあたり多大なるご指導とご協力を頂いた大阪大学神経内科学教室の望月秀樹先生、池中建介先生、同大学遺伝学教室の吉森保先生、金沢大学微粒子システム研究室瀬戸章文先生ならびに両研究室の共同研究者の皆様に深く御礼申し上げます。またこのような光栄な場での執筆の機会を与えていただきました日本神経学会出版・広報委員会の諸先生方ならびに関係者の皆様に心より御礼申し上げます。
1) Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature, 388(6645), 839–840 (1997).
2) Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science, 276(5321), 2045–2047 (1997).
3) Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging, 24(2), 197–211 (2003). http://www.sciencedirect.com/science/article/pii/S0197458002000659
4) Karpowicz RJ Jr., Trojanowski JQ, Lee VM. Transmission of alpha-synuclein seeds in neurodegenerative disease: recent developments. Lab Invest, 99(7), 971–981 (2019). http://www.ncbi.nlm.nih.gov/pubmed/30760864
5) Ahmed I, Liang Y, Schools S, Dawson VL, Dawson TM, Savitt JM. Development and characterization of a new Parkinson’s disease model resulting from impaired auto phagy. J Neurosci, 32(46), 16503–16509 (2012).
6) Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, Tan M, Kia DA, Noyce AJ, Xue A, Bras J, Young E, von Coelln R, Simón-Sánchez J, Schulte C, Sharma M, Krohn L, Pihlstrøm L, Siitonen A, Iwaki H, Leonard H, Faghri F, Gibbs JR, Hernandez DG, Scholz SW, Botia JA, Martinez M, Corvol JC, Lesage S, Jankovic J, Shulman LM, Sutherland M, Tienari P, Majamaa K, Toft M, Andreassen OA, Bangale T, Brice A, Yang J, Gan-Or Z, Gasser T, Heutink P, Shulman JM, Wood NW, Hinds DA, Hardy JA, Morris HR, Gratten J, Visscher PM, Graham RR, Singleton AB, Adarmes-Gómez AD, Aguilar M, Aitkulova A, Akhmetzhanov V, Alcalay RN, Alvarez I, Alvarez V, Bandres-Ciga S, Barrero FJ, Bergareche Yarza JA, Bernal-Bernal I, Billingsley K, Blauwendraat C, Blazquez M, Bonilla-Toribio M, Botía JA, Boungiorno MT, Bras J, Brice A, Brockmann K, Bubb V, Buiza-Rueda D, Cámara A, Carrillo F, Carrión-Claro M, Cerdan D, Chelban V, Clarimón J, Clarke C, Compta Y, Cookson MR, Corvol JC, Craig DW, Danjou F, Diez-Fairen M, Dols-Icardo O, Duarte J, Duran R, Escamilla-Sevilla F, Escott-Price V, Ezquerra M, Faghri F, Feliz C, Fernández M, Fernández-Santiago R, Finkbeiner S, Foltynie T, Gan-Or Z, Garcia C, García-Ruiz P, Gasser T, Gibbs JR, Gomez Heredia MJ, Gómez-Garre P, González MM, Gonzalez-Aramburu I, Guelfi S, Guerreiro R, Hardy J, Hassin-Baer S, Hernandez DG, Heutink P, Hoenicka J, Holmans P, Houlden H, Infante J, Iwaki H, Jesús S, Jimenez-Escrig A, Kaishybayeva G, Kaiyrzhanov R, Karimova A, Kia DA, Kinghorn KJ, Koks S, Krohn L, Kulisevsky J, Labrador-Espinosa MA, Leonard HL, Lesage S, Lewis P, Lopez-Sendon JL, Lovering R, Lubbe S, Lungu C, Macias D, Majamaa K, Manzoni C, Marín J, Marinus J, Marti MJ, Martinez M, Martínez Torres I, Martínez-Castrillo JC, Mata M, Mencacci NE, Méndez-del-Barrio C, Middlehurst B, Mínguez A, Mir P, Mok KY, Morris HR, Muñoz E, Nalls MA, Narendra D, Noyce AJ, Ojo OO, Okubadejo NU, Pagola AG, Pastor P, Perez Errazquin F, Periñán-Tocino T, Pihlstrom L, Plun-Favreau H, Quinn J, R’Bibo L, Reed X, Rezola EM, Rizig M, Rizzu P, Robak L, Rodriguez AS, Rouleau GA, Ruiz-Martínez J, Ruz C, Ryten M, Sadykova D, Scholz SW, Schreglmann S, Schulte C, Sharma M, Shashkin C, Shulman JM, Sierra M, Siitonen A, Simón-Sánchez J, Singleton AB, Suarez-Sanmartin E, Taba P, Tabernero C, Tan MX, Tartari JP, Tejera-Parrado C, Toft M, Tolosa E, Trabzuni D, Valldeoriola F, van Hilten JJ, Van Keuren-Jensen K, Vargas-González L, Vela L, Vives F, Williams N, Wood NW, Zharkinbekova N, Zharmukhanov Z, Zholdybayeva E, Zimprich A, Ylikotila P, Shulman LM, von Coelln R, Reich S, Savitt J, Agee M, Alipanahi B, Auton A, Bell RK, Bryc K, Elson SL, Fontanillas P, Furlotte NA, Huber KE, Hicks B, Jewett EM, Jiang Y, Kleinman A, Lin KH, Litterman NK, McCreight JC, McIntyre MH, McManus KF, Mountain JL, Noblin ES, Northover CAM, Pitts SJ, Poznik GD, Sathirapongsasuti JF, Shelton JF, Shringarpure S, Tian C, Tung J, Vacic V, Wang X, Wilson CH, Anderson T, Bentley S, Dalrymple-Alford J, Fowdar J, Gratten J, Halliday G, Henders AK, Hickie I, Kassam I, Kennedy M, Kwok J, Lewis S, Mellick G, Montgomery G, Pearson J, Pitcher T, Sidorenko J, Silburn PA, Vallerga CL, Visscher PM, Wallace L, Wray NR, Xue A, Yang J, Zhang F; 23andMe Research Team; System Genomics of Parkinson’s Disease Consortium; International Parkinson’s Disease Genomics Consortium. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol, 18(12), 1091–1102 (2019).
7) Dehay B, Bové J, Rodríguez-Muela N, Perier C, Recasens A, Boya P, Vila M. Pathogenic lysosomal depletion in Parkinson’s disease. J Neurosci, 30(37), 12535–12544 (2010).
8) Cuervo AM, Stafanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science, 305(5688), 1292–1295 (2004).
9) Nakamura S, Oba M, Suzuki M, Takahashi A, Yamamuro T, Fujiwara M, Ikenaka K, Minami S, Tabata N, Yamamoto K, Kubo S, Tokumura A, Akamatsu K, Miyazaki Y, Kawabata T, Hamasaki M, Fukui K, Sango K, Watanabe Y, Takabatake Y, Kitajima TS, Okada Y, Mochizuki H, Isaka Y, Antebi A, Yoshimori T. Suppression of autophagic activity by Rubicon is a signature of aging. Nat Commun, 10(1), 847 (2019). http://www.ncbi.nlm.nih.gov/pubmed/30783089
10) Oe Y, Kakuda K, Yoshimura SI, Hara N, Hasegawa J, Terawaki S, Kimura Y, Ikenaka K, Suetsugu S, Mochizuki H, Yoshimori T, Nakamura S. PACSIN1 is indispensable for amphisome-lysosome fusion during basal autophagy and subsets of selective autophagy. PLoS Genet, 18(6), 1–27 (2022). http://dx.doi.org/10.1371/journal.pgen.1010264
11) Domingues R, Sant’Anna R, da Fonseca ACC, Robbs BK, Foguel D, Outeiro TF. Extracellular alpha-synuclein: Sensors, receptors, and responses. Neurobiol Dis, 168, 105696 (2021). https://doi.org/10.1016/j.nbd.2022.105696
12) Jiang P, Gan M, Yen SH, McLean PJ, Dickson DW. Impaired endo-lysosomal membrane integrity accelerates the seeding progression of alpha-synuclein aggregates. Sci Rep, 7(1), 7690 (2017). https://www.ncbi.nlm.nih.gov/pubmed/28794446
13) Kakuda K, Ikenaka K, Kuma A, Doi J, Aguirre C, Wang N, Ajiki T, Choong CJ, Kimura Y, Badawy SMM, Shima T, Nakamura S, Baba K, Nagano S, Nagai Y, Yoshimori T, Mochizuki H. Lysophagy protects against propagation of a-synuclein aggregation through ruptured lysosomal vesicles. Proc Natl Acad Sci USA, 121(1), e2312306120 (2024).
14) Navarro-Romero A, Fernandez-Gonzalez I, Riera J, Montpeyo M, Albert-Bayo M, Lopez-Royo T, Castillo-Sanchez P, Carnicer-Caceres C, Arranz-Amo JA, Castillo-Ribelles L, Pradas E, Casas J, Vila M, Martinez-Vicente M. Lysosomal lipid alterations caused by glucocerebrosidase deficiency promote lysosomal dysfunction, chaperone-mediated-autophagy deficiency, and alpha-synuclein pathology. NPJ Parkinsons Dis, 8(1), 126 (2022).
15) Shi L, Wu X, Danesh Yazdi M, Braun D, Abu Awad Y, Wei Y, Liu P, Di Q, Wang Y, Schwartz J, Dominici F, Kioumourtzoglou MA, Zanobetti A. Long-term effects of PM2·5 on neurological disorders in the American Medicare population: a longitudinal cohort study. Lancet Planet Health, 4(12), e557–e565 (2020). http://dx.doi.org/10.1016/S2542-5196(20)30227-8
16) Papadopoulos C, Kravic B, Meyer H. Repair or Lysophagy: Dealing with Damaged Lysosomes. J Mol Biol, 432(1), 231–239 (2020). https://doi.org/10.1016/j.jmb.2019.08.010
17) Radulovic M, Schink KO, Wenzel EM, Nahse V, Bongiovanni A, Lafont F, Stenmark H. ESCRT-mediated lysosome repair precedes lysophagy and promotes cell survival. EMBO J, 37(21), e99753 (2018). http://www.ncbi.nlm.nih.gov/pubmed/30314966
18) Maejima I, Takahashi A, Omori H, Kimura T, Takabatake Y, Saitoh T, Yamamoto A, Hamasaki M, Noda T, Isaka Y, Yoshimori T. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J, 32(17), 2336–2347 (2013). https://www.ncbi.nlm.nih.gov/pubmed/23921551
19) Chen JJ, Nathaniel DL, Raghavan P, Nelson M, Tian R, Tse E, Hong JY, See SK, Mok SA, Hein MY, Southworth DR, Grinberg LT, Gestwicki JE, Leonetti MD, Kampmann M. Compromised function of the ESCRT pathway promotes endolysosomal escape of tau seeds and propagation of tau aggregation. J Biol Chem, 294(50), 18952–18966 (2019).
20) Falcon B, Noad J, McMahon H, Randow F, Goedert M. Galectin-8-mediated selective autophagy protects against seeded tau aggregation. J Biol Chem, 293(7), 2438–2451 (2018). http://dx.doi.org/10.1074/jbc.M117.809293

This page was created on 2024-06-24T08:44:01.408+09:00
This page was last modified on 2024-07-23T09:32:39.000+09:00
このサイトは(株)国際文献社によって運用されています。