リキッドバイオプシーによる神経変性疾患バイオマーカー開発
大阪大学大学院医学系研究科神経内科学
発行日:2019年6月30日Published: June 30, 2019
© 2019 日本神経化学会© 2019 The Japanese Society for Neurochemistry
アルツハイマー病、パーキンソン病、筋萎縮性側索硬化症といった神経変性疾患は、緩徐進行性に特定の部位の神経脱落と、その部位の障害に由来する神経徴候を呈する疾患の総称である。長らく原因は不明であったが、一部の家族性神経変性疾患患者の分子遺伝学的な解析や免疫組織学的手法の発展により、神経変性疾患はそれぞれ特定の蛋白質の凝集・蓄積により起こされることが明らかになり1, 2, 3)、プロテイノパチーという概念が今では広く認知されている。その後、生前の臨床像と剖検の病理学的な比較検討が進み、臨床的診断基準が確立されてきた。しかしながら、未だに確定診断は死後の病理組織による確認を要し、生前の診断の確度は十分ではない。
実はこの点が癌を始めとする生検が可能な疾患と、中枢神経の変性疾患の大きな違いであり、侵襲性や合併症の問題から脳や脊髄の生検がほぼ不可能である。これは治療方法がない時代においては大きな問題ではなかったが、凝集蛋白質の除去や神経保護薬など様々な疾患修飾療法が開発されてきている現在では極めて重要な点である。超早期の治療が治療薬開発のカギとなってきており、臨床症状が十分に進行して初めて確定できる臨床診断では治療開始が遅すぎる可能性がある。
より早期の診断のために、場合によっては発症前の診断を可能にするための技術として脳内の病理病態を反映するバイオマーカーの開発、中でも生体試料から凝集蛋白質の検出技術「リキッドバイオプシー」の開発が近年大きな高まりを見せている。その中で見えてきた新しい病態解明の可能性、今後の治療の可能性などについて、我々の最新の研究結果を交えて、特にパーキンソン病に焦点を当てて概説する。
パーキンソン病(PD)は緩徐進行性の神経変性疾患で、安静時振戦・筋強剛・無動・姿勢反射障害などの運動徴候を主徴とする。近年では、運動障害に追加して、認知機能障害・嗅覚障害・自律神経障害といった、非運動障害の重要性が広く認知されるようになってきた。PDの病理学的特徴は、中脳黒質のドパミン産生ニューロンなど様々な神経細胞において観察されるレビー小体と呼ばれる蛋白質凝集物の封入体の形成であり、その主要構成成分がαシヌクレイン(αSyn)であることが知られている4, 5)。図1に一般的なPDの病理的・臨床的経過をまとめた。αSynの蓄積から始まり、徐々に神経細胞の脱落が起こり、まず非運動症状が出現したのちに運d脳障害が出現する(図1)PDの主な臨床診断は、運動障害に着目したものが主体となっており、運動症状がある程度進んで初めて臨床的に診断される。一方で、神経細胞に着目すると、運動機能障害の出現時にはすでに30~80%の中脳黒質細胞の脱落が認められるという報告もある6)。αSyn凝集阻害薬などの疾患修飾療法の開発には、運動症状発症前に病態を捉えて治療を開始する事が望ましいことがわかる。そのためには現状の診断基準による運動症状出現前の前駆症状期(prodromal PD)または発症前期(preclinical PD)を捉える必要がある。それを可能にするために各種バイオマーカーの開発が必須であり、また運動症状発症前にいかにして患者を見つけられるかがカギになる。

どのようにしてprodromal PDの患者を捉えるかが課題となると前述したが、健康診断ベースでのスクリーニングは安価で簡便で高い感度を持つ検査が要求され、現状ではそれを満たすバイオマーカーは存在しない。しかし、これまでのパーキンソン病の横断的な研究から、運動症状が出現する時期(中脳黒質—線条体系の障害を受ける時期)より以前に、PD患者はいくつかの特徴的な臨床像を高頻度に呈することがわかってきた7)。特に特徴的な症状としては、レム睡眠行動異常症、嗅覚障害、便秘、泌尿器障害(排尿障害、インポテンツ)などがあげられる。運動症状がない患者で、これらの前駆症状診断基準を用いると、80%の確度で将来のPDを診断できる。下記に、代表的なレム睡眠行動異常症と、嗅覚障害についてさらに概説する。
通常レム睡眠時には全身の骨格筋の緊張が低下しているが、何らかの原因で筋緊張の抑制が障害されるために、夢で見たことをそのまま行動に移してしまう疾患である。RBDはパーキンソン病やその他のαSynが蓄積する疾患において高頻度に認められる。PDでは33~60%程度の患者にRBDが認められると報告されている8)。またRBDはprodromal PDにおいても高頻度に認められることが知られており、PDの早期診断マーカーとして期待されている。RBDの診断後の縦断的追跡調査では、10年間で80%程度の患者がパーキンソン病等を発症すると報告されている9)。現在、国立精神・神経医療研究センターや我々阪大神経内科など国内5施設において、RBD患者を追跡調査することで、パーキンソン病の発症前マーカーの探索を行うプロジェクト、J-PPMI(Japan-Parkinson’s Progressive Markers Initiative)を展開している。
PDでは病早期、または発症前から嗅覚低下がみられることが多い。嗅覚障害が早期から起こることは、Braakらの横断的な病理学的解析により、PDの中枢初期病変は嗅球あるいは迷走神経背側核からはじまるという報告にも裏付けされている10)。MSAやPSPなどでは障害されにくいため、鑑別診断に有用である。
また、近年PDにおける重度嗅覚低下が後の認知症発症の予測になりうる事が報告された11)。つまり、PDがさらに進行して、認知症を合併するかどうかを予測できる臨床マーカーといえる。これをうけて2013年からDonepezil Application for Severe Hyposmic Parkinson Disease(DASH-PD)研究が始まっている。この研究は認知機能障害がなく重度嗅覚障害を有するPD患者を、ドネペジル投与群とプラセボ投与群の2群に分けて将来の認知症発症予防が可能かどうかを調べている。
前述の臨床的マーカーの活用により、将来的にPDを発症する人を絞り込むことができ、期待値を上げたうえで生化学的にαSynの検出、モニタリングをしていくことが可能となる。これまでαSyn量を推測する確立された生化学的バイオマーカーは存在しないが、現在αSyn蓄積を検出するバイオ—マーカー開発は世界的に活発に行われている。代表的な方法は、ELISA法と、アミロイド蛋白質増幅法の二種類がある。
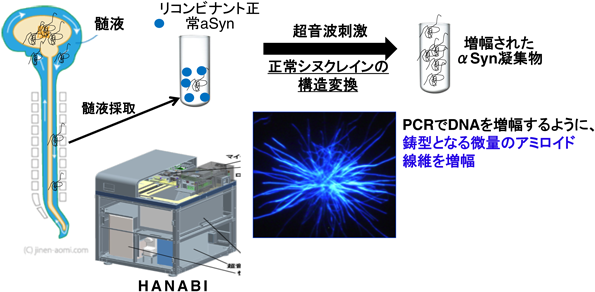
京都府立医科大学の徳田らは、αSynのELISA開発を進めてきており、αSynオリゴマーを特異的に検出するsingle-antibody sandwich ELISA法を用いてPD患者では髄液中の正常なαSynは低下し、αSyn凝集体が増加する事を見出した12)。さらに最近では、より高感度に髄液中のαSynを検出するRT-QuIC法が開発されており、中枢神経系におけるαSynを定量評価する方法が開発され注目されている13)。我々の研究室においても、RT-QuICをベースに超音波を用いた新しい髄液中シヌクレイン凝集体検出装置、HANABI(HANdai Amyloid Burst Inducer)を開発している。次項以降にアミロイド蛋白質増幅、検出の手法と、PD患者における結果を概説していきたい。
プリオン蛋白質やαSynなどアミロイド原生蛋白質は、異常なクロスβシート構造を特徴とするフィブリルを形成する。その大きな特徴として、正常型構造を有する蛋白質分子を自身と同じ異常型クロスβシート構造に変換する蛋白質間の構造伝播がある(図2)。この蛋白質間構造伝播は、異常蛋白質が細胞から細胞に移動することで、細胞間伝播につながり、神経変性疾患の進行を規定している。
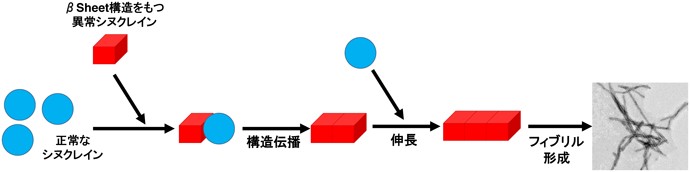
アミロイド蛋白質増幅法は、この構造伝播の性質を利用した微量アミロイド蛋白質の検出法で、これまでPMCA法、RT-QuIC法、そして我々のHANABI法が開発されてきた14)。どの方法においても、正常なシヌクレイン(大腸菌由来のリコンビナント蛋白質)に異常蛋白質を含む試料(患者脊髄液など)を添加し、刺激を加えて反応促進することで異常蛋白質を検出できるレベルまで増幅する(図3)。PMCA法とHANABI法は超音波を用いて刺激し、RT-QuIC法は攪拌を用いる。検出はPMCA法はプロテアーゼK耐性で評価し、RT-QuIC法とHANABI法はアミロイド線維に結合し蛍光を発するチオフラビンT(ThT)を用いる(図4)。
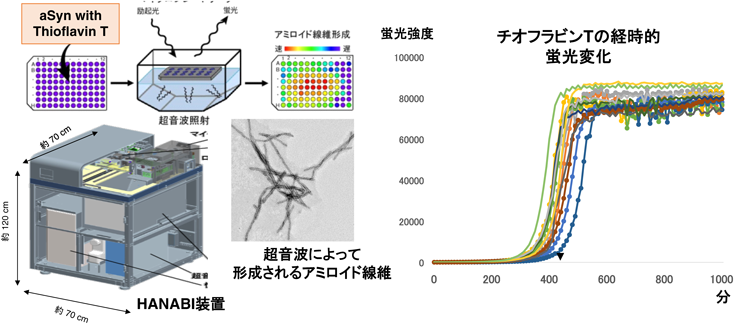
特にRT-QuIC法とHANABI法においては、ThTの蛍光変化を追跡することができ、正常シヌクレインの構造が変換されていく速度を定量評価できる。元の試料に含まれる異常シヌクレインが多ければ凝集過程は早くなることから、試料中の凝集量の評価をすることができる。例えば、人工的に作成したαSynフィブリルを添加することで、濃度依存性に凝集を加速することができ、凝集曲線のT(1/2)時間を測り定量評価できる(図5)。この系を用いて、我々は、培養細胞において細胞内にαSynの凝集体ができる系を用いて、細胞内から細胞外に放出されるαSyn凝集物の評価に成功した。
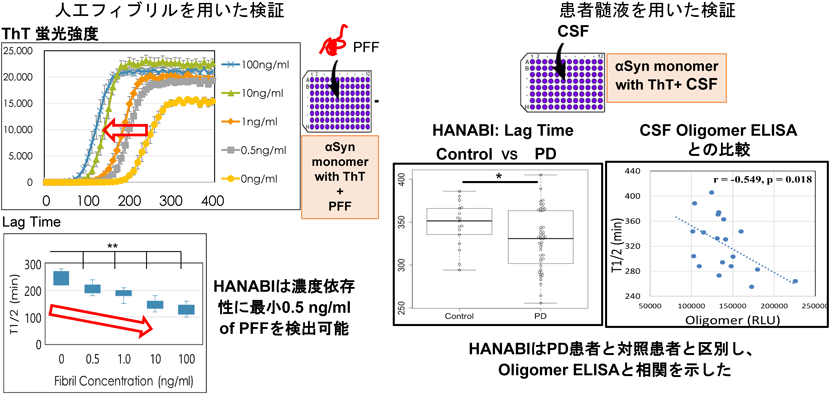
まず我々は、PD患者と対照患者を比較して、PD患者髄液は有意に反応を促進することを示した(図5)。さらにPD患者においては、京都府立医大徳田らによるオリゴマーELISAによるαSyn凝集体定量法との比較検討をしたところ、優位な相関を示した(図5)。さらに、様々な臨床スコアや画像検査との相関を調べたところ、心臓交感神経の脱落を評価するMIBG心筋シンチグラフィーの結果と相関を示した。これまでの病理学的な解析結果から、PD患者における心臓交感神経の脱落は、心臓交感神経節におけるαSynの凝集蓄積が原因であることが知られており、同時に脳内のαSyn病理と非常に関連が深いことが知られている15)。つまり、HANABIによる髄液αSynの凝集体の量的な評価は、脳内のαSyn変化を見ている可能性があることを間接的に証明したことになり、意義が深い。
最初に述べたように、神経変性疾患患者において脳から生検をすることは不可能である。しかし、脳内で蓄積している蛋白質を髄液中から検出できる可能性が示されてきている。それを用いることで病態の解明につながる可能性がある。
例えば、αSynが蓄積する疾患はPD以外にもグリア細胞に主に蓄積し、小脳失調や自律神経失調など多様な症状を呈して急速に進行する多系統萎縮症(MSA)という疾患がある。その違いを規定するものはなにか、長年の大きな疑問であるが、その違いが凝集体の構造の違い(構造多型)に起因する可能性が示唆されている。つまりαSynの凝集の過程において異なる構造の凝集物ができ、その構造によって毒性、細胞選択性、ひいては症状を変えるという仮説である16, 17, 18)。この仮説を検証する際にHANABIを始めとしたアミロイド蛋白質増幅法が有用である。つまり、PD患者、MSA患者の髄液をHANABIなどを用いて構造を保持したまま特異的に増幅し、その高次構造を解析することで脳内に蓄積しているαSyn凝集物の性質の違いを再現する試みである(図6)。

今後の神経変性疾患のバイオマーカーの大きな潮流は、このように凝集している蛋白質の量的な評価と、質的な評価を実現することで、これまで死後脳でしか評価ができなかった凝集蛋白質の評価を生前に可能にすることである。くしくもそれは、癌領域において注目を集めるリキッドバイオプシーとしての意義と極めて似ていて、これまでバイオプシーができないことで研究の進展に壁のあった神経変性疾患においても大きなブレイクスルーとなる。今後このバイオマーカーが様々な角度から臨床的意義、病理との比較検討が進められていき、さらに新薬開発の治験デザインに組み込まれていくことで、世界的に難航している神経変性疾患の根本的な治療法開発の大きな推進力になると信じて開発を進めている。
HANABIの開発におきまして、多くのご支援とご指導を頂きました大阪大学蛋白質研究所後藤祐児教授、およびその研究室の皆様に感謝いたします。さらにこのような執筆の機会をいただきました澤本和延教授および神経化学会の編集部の皆様に感謝申し上げます。また、本稿は極めて臨床的な内容で、皆様に興味を持って読んでいただけるか心配しながら執筆いたしました。最後までお読みくださいました皆様に深く御礼申し上げます。
1) Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, Hung WY, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak-Vance MA, Haines J, Rouleau GA, Gusella JS, Horvitz HR, Brown RH. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature, 362(6415), 59–62 (1993).
2) Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin JF, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Da Silva HA, Haines JL, Perkicak-Vance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, St George-Hyslop PH. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature, 375(6534), 754–760 (1995).
3) Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science, 276(5321), 2045–2047 (1997).
4) Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature, 388(6645), 839–840 (1997).
5) Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol, 4(2), 160–164 (2002).
6) Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: Substantia nigra regional selectivity. Brain, 114(Pt 5), 2283–2301 (1991).
7) Berg D, Postuma RB, Adler CH, Bloem BR, Chan P, Dubois B, Gasser T, Goetz CG, Halliday G, Joseph L, Lang AE, Liepelt-Scarfone I, Litvan I, Marek K, Obeso J, Oertel W, Olanow CW, Poewe W, Stern M, Deuschl G. MDS research criteria for prodromal Parkinson’s disease. Mov Disord, 30(12), 1600–1611 (2015).
8) Boeve BF, Silber MH, Saper CB, Ferman TJ, Dickson DW, Parisi JE, Benarroch EE, Ahlskog JE, Smith GE, Caselli RC, Tippman-Peikert M, Olson EJ, Lin SC, Young T, Wszolek Z, Schenck CH, Mahowald MW, Castillo PR, Del Tredici K, Braak H. Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease. Brain, 130(Pt 11), 2770–2788 (2007).
9) Iranzo A, Tolosa E, Gelpi E, Molinuevo JL, Valldeoriola F, Serradell M, Sanchez-Valle R, Vilaseca I, Lomeña F, Vilas D, Lladó A, Gaig C, Santamaria J. Neurodegenerative disease status and post-mortem pathology in idiopathic rapid-eye-movement sleep behaviour disorder: An observational cohort study. Lancet Neurol, 12(5), 443–453 (2013).
10) Braak E, Sandmann-Keil D, Rüb U, Gai WP, de Vos RA, Steur EN, Arai K, Braak H. alpha-synuclein immunopositive Parkinson’s disease-related inclusion bodies in lower brain stem nuclei. Acta Neuropathol, 101(3), 195–201 (2001).
11) Baba T, Kikuchi A, Hirayama K, Nishio Y, Hosokai Y, Kanno S, Hasegawa T, Sugeno N, Konno M, Suzuki K, Takahashi S, Fukuda H, Aoki M, Itoyama Y, Mori E, Takeda A. Severe olfactory dysfunction is a prodromal symptom of dementia associated with Parkinson’s disease: A 3 year longitudinal study. Brain, 135(Pt 1), 161–169 (2012).
12) Tokuda T, Qureshi MM, Ardah MT, Varghese S, Shehab SA, Kasai T, Ishigami N, Tamaoka A, Nakagawa M, El-Agnaf OM. Detection of elevated levels of alpha-synuclein oligomers in CSF from patients with Parkinson disease. Neurology, 75(20), 1766–1772 (2010).
13) Shahnawaz M, Tokuda T, Waragai M, Mendez N, Ishii R, Trenkwalder C, Mollenhauer B, Soto C. Development of a Biochemical Diagnosis of Parkinson Disease by Detection of alpha-Synuclein Misfolded Aggregates in Cerebrospinal Fluid. JAMA Neurol, 74(2), 163–172 (2017).
14) Parnetti L, Gaetani L, Eusebi P, Paciotti S, Hansson O, El-Agnaf O, Mollenhauer B, Blennow K, Calabresi P. CSF and blood biomarkers for Parkinson’s disease. Lancet Neurol, (2019).
15) Orimo S, Amino T, Itoh Y, Takahashi A, Kojo T, Uchihara T, Tsuchiya K, Mori F, Wakabayashi K, Takahashi H. Cardiac sympathetic denervation precedes neuronal loss in the sympathetic ganglia in Lewy body disease. Acta Neuropathol, 109(6), 583–588 (2005).
16) Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, Madiona K, Olieric V, Böckmann A, Meier BH, Melki R. Structural and functional characterization of two alpha-synuclein strains. Nat Commun, 4(1), 2575 (2013).
17) Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, Van den Haute C, Melki R, Baekelandt V. alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature, 522(7556), 340–344 (2015).
18) Peelaerts W, Bousset L, Baekelandt V, Melki R. α-Synuclein strains and seeding in Parkinson’s disease, incidental Lewy body disease, dementia with Lewy bodies and multiple system atrophy: Similarities and differences. Cell Tissue Res, 373(1), 195–212 (2018).

This page was created on 2019-05-15T13:44:05.051+09:00
This page was last modified on 2019-06-17T18:16:47.000+09:00
このサイトは(株)国際文献社によって運用されています。