マイクログリアによる補体依存的なシナプス刈り込み
国立精神・神経医療研究センター 神経研究所 疾病研究第二部
発行日:2025年6月30日Published: June 30, 2025
© 2025 日本神経化学会© 2025 The Japanese Society for Neurochemistry
脳のグリア細胞の一種であるマイクログリアは免疫細胞としての機能を有し、脳内の異物や病原体などの除去や炎症性メディエーターの産生・放出を行う。近年、マイクログリアは外傷や感染による炎症時だけではなく、健常脳においても様々な役割を果たすことが明らかとなっている。その一つが脳内で生じた不要物(死細胞や凝集タンパク質など)の貪食による除去である。これらが除去されずに脳内に残存すると、活性酸素種や種々の炎症性メディエーターの放出を介して組織の損傷を引き起こすため、マイクログリアによる貪食作用は脳機能の恒常性維持に重要であると考えられてきた1–4)。他の臓器における免疫細胞と同様、マイクログリアは除去対象に結合した補体を目印として貪食を行う。さらに、このような補体依存的な貪食作用は、神経細胞間の情報伝達の場であるシナプスに対しても発揮されることが明らかとなってきた5)。そして、様々な状況における補体依存的なシナプス貪食が検証された結果、この機構が正常な脳機能の発揮や脳疾患の発症に重要である可能性が示されている。本稿では、まず、健常脳および疾患脳における補体依存的なシナプス貪食について概説する。その後、マイクログリアがシナプスを貪食するメカニズムに関する我々の最新の研究成果を紹介する。
シナプスは神経細胞間の情報伝達の場であり、プレシナプスとポストシナプスの適切な結合による神経回路の構築が正常な脳機能の発揮に重要である。シナプスは生涯にわたって形成と消失を繰り返すことで、神経回路の再編成を実現している。特に発達期においては、シナプス密度が大きく変化することが、ヒトやげっ歯類の死後脳の観察により明らかとなっている。具体的には、シナプス密度が生後から発達初期にかけて増加した後、青年期に減少し壮年期にはほぼ一定となる6)。この発達期におけるシナプス密度の減少はシナプス刈り込みと呼ばれ、不要なシナプスが消失し残存したシナプスが形態的・機能的に成熟することが、神経回路の精緻化に重要であると考えられてきた。実際に、自閉症スペクトラム症(autism spectrum disorders; ASD)や統合失調症といった神経発達障害の脳では、健常脳と比較してそれぞれシナプス密度の増加と減少が確認されている7)。
発達期のシナプス刈り込みにマイクログリアの貪食能が関与する可能性は、2011年にPaolicelliらによって初めて示された8)。この研究では、発達期のマウスを用いて免疫組織化学を行い、海馬CA1野においてマイクログリアの内部に興奮性プレシナプスと興奮性ポストシナプスが取り込まれた様子を捉えた。ほぼ同時期にSchaferらは、発達期のマウスの網膜–外側膝状体経路において、マイクログリアによるシナプス貪食が補体経路を担うC1qやC3によって制御されることを示した9)。C3やその受容体であるCR3をknockout (KO)したマウスでは、マイクログリアに内包されるプレシナプスの量が減少していた。また、薬理学的実験によって、補体は神経活動が相対的に低いプレシナプスをタグ付けすることが分かった。これらのデータから、マイクログリアがCR3を介して活動の弱いシナプス上のC3を認識し、そのシナプスを貪食することが示唆された(図1)。
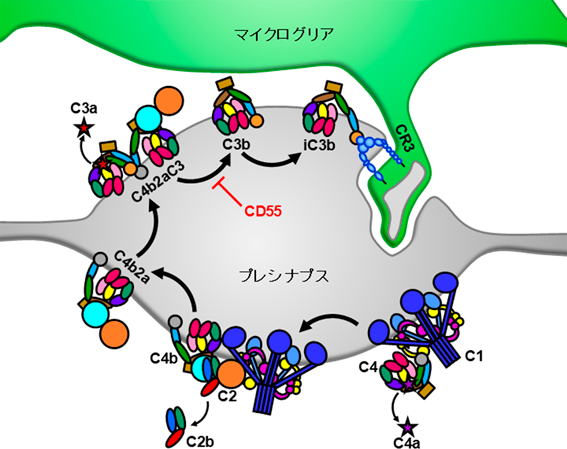
C1qを起点とする古典的補体活性化経路が、マイクログリアによるシナプス貪食を誘導するまでのシグナル伝達。マイクログリアはCR3を構成するインテグリンCD11bを介してiC3bを認識し、シナプスを貪食する。図は文献10より改変。
発達期の神経回路形成に加えて、生理学的な神経回路可塑性に対する補体依存的なシナプス貪食の関与も示唆されている。記憶の整理や定着を促進することが知られている睡眠時には、マイクログリアの活性化(細胞体肥大化、CR3発現量増加)と補体の発現量増加が確認された11)。なお、同じタイミングで、C3が結合したシナプスが増加し、その一部はマイクログリアにより貪食されていたことから、シナプスの日内変動にマイクログリアによる貪食が寄与することが示唆された。また、マイクログリアが記憶の忘却に関わる可能性も報告された12)。マウスに対する文脈的恐怖条件づけにおいて、学習成立後に、CSF1R阻害剤のPLX3397を投与することによってマイクログリア(PLX3397は他のマクロファージや単球などにも影響を及ぼすことに注意されたい)を除去すると記憶の保持期間が延長した。この研究では記憶の忘却にシナプス刈り込みが関与する可能性を考え、C3の活性化を阻害するCD55を学習時に活動した神経細胞に発現させ、補体依存的なシナプス貪食を阻害した。その結果、CD55の発現により文脈的恐怖条件づけの記憶保持期間が延長したこと、マイクログリアによるCD55発現神経細胞の貪食が減少したことから、補体依存的なシナプス貪食が記憶学習を制御する可能性が示された。
発達期のシナプス貪食が正常に行われないと、脳機能の発達にどのような影響が生じるのだろうか。C1qをKOしたマウスの皮質では、興奮性シナプスの密度や機能的結合性の増加が見られ、発達期のシナプス刈り込みが不全となる可能性が示された13)。また、C1q KOマウスではてんかん様発作の脳波が観察されたことから、C1qのKOはシナプス貪食を抑制することで神経回路の過剰興奮を引き起こすことが示唆された。先に述べたように、ASD脳では発達期のシナプス刈り込みが障害される結果、シナプス密度が増加することが示唆されている。また、ASDモデル動物ではマイクログリアによるシナプス貪食が不全であるという報告も多い。さらに、ASD患者の前頭葉ではC3の発現レベルが低下すること、マウス脳においてC3をknockdownするとASD様行動(社会性低下、常同行動)が顕在化することから14)、補体経路の異常とASD発症との関連性が伺える。しかしながら、ASD発症と補体依存的なシナプス貪食異常との関連性を直接的に検証した例はない。統合失調症患者の死後脳では、皮質におけるシナプス密度の低下が確認されており、過剰なシナプス刈り込みが統合失調症の発症原因として示唆されてきた。Sellgrenらは、マイクログリア依存的なシナプス貪食のin vitroモデルを構築し、この可能性を検証した15)。そして、統合失調症患者由来の神経細胞やシナプトソームがより貪食されやすいこと、患者由来のマイクログリア様細胞の貪食能がより高いことを示した。さらに、統合失調症リスク遺伝子であるC4が、神経細胞への補体付着やシナプス除去に関与することを発見した。
アルツハイマー病(Alzheimer’s disease; AD)をはじめとする神経変性疾患の脳では、しばしばシナプスの脱落が見られる。ADの他、様々な神経変性疾患のモデル動物を用いた検証により、脳内に凝集したタンパク質や炎症、組織損傷がマイクログリアを活性化することで過剰なシナプス除去を引き起こすことが明らかとなっている。また、こうした疾患の多くでは、補体分子の過剰な発現増加や活性化が生じており、発達期のシナプス除去メカニズムが再び活性化されることが神経変性疾患の原因であることが示唆されている。例えば、ADモデルマウスでは、C1qがポストシナプスをタグ付けし、マイクログリアによるポストシナプス貪食を促進する結果、シナプス密度が低下することが示されている16, 17)。C1qによるシナプスのタグ付けが促進されるメカニズムとして、シナプスに付着したAβオリゴマーが直接的もしくは間接的にC1qと結合する可能性が考えられている。また、前頭側頭葉変性症のリスク遺伝子でprogranulinをコードするGrnをKOしたマウスにおいても、C1qaやC3の発現が増加する。その結果、C1qによるプレシナプスのタグ付けとマイクログリアによるプレシナプスの貪食が促進された18)。
我々の論文を含む最新の研究では、マイクログリアによる補体依存的な抑制性シナプスの貪食が神経回路の興奮性を上昇させ、てんかんの発症や悪化を促進する可能性が示された19, 20)。後述するが、我々の研究では抑制性シナプス特異的に補体によるタグ付けが増加するメカニズムにまで言及した。
第1、2章で紹介したように、過去15年ほどの間に、マイクログリアによる補体依存的なシナプス貪食に関する研究が精力的に行われてきた。しかしその一方で、この現象のメカニズムには、いまだ解明されていない重要な点が残されていると筆者は考えている。とりわけ注目すべきは、マイクログリアが本当にシナプス部分のみを選択的に貪食しているのか、すなわち神経細胞(軸索)はシナプス貪食後でも無傷で保たれているのか、という点である。これまで、マイクログリアによる選択的なシナプス除去や、神経細胞へのダメージを伴わない貪食は、マイクログリアによる神経回路可塑性の制御を論じるうえで前提とされてきたが、実験的にこれを証明することは容易ではなかった。
近年、観察技術の進歩により、海馬切片培養を用いた研究において、マイクログリアがプレシナプス構造(ブートン)を取り込む様子が観察されるようになった21)。この研究は、マイクログリアによるシナプス構造のトロゴサイトーシス(標的細胞の一部をかじり取る貪食様式)という概念を提示した点で重要である。しかしながら、同研究でマイクログリアに取り込まれた構造体には、シナプス特異的なタンパク質が含まれていたかどうかは確認されておらず、取り込まれた構造が本当に軸索と連続性を保った“機能的シナプス”であったのかどうかも不明であった。
また、Limらによる研究では、pH安定型蛍光タグ(Synaptophysin-pHtdGFP)を用いてマイクログリアへの蛍光蓄積が観察された22)。彼らはアフリカツメガエルを用いたin vivoタイムラプスイメージングを実現したが、この研究もリアルタイムで分子同定されたシナプス構造の取り込みを直接可視化したものではなかった。
我々は、マイクログリアが生きた神経細胞からシナプスのみを選択的に貪食する証拠をつかむべく、高い時空間解像度でマイクログリアによるシナプス貪食を観察する培養系を構築した。従来、培養下のマイクログリアではその特徴である細長く分岐した多数の突起が失われることが問題となっていた。そこで、培養条件(培地組成や培養日数など)を検討し、この問題点を克服した(図2(A))。そして、トランスジェニックマウスおよびアデノ随伴ウィルスの利用により、マイクログリア(CX3CR1-GFP/+マウス)、シナプス小胞マーカー(Synaptophysin-mCherry)、神経細胞軸索膜(membrane-targeted iRFP)を可視化した。タイムラプスイメージングの結果、マイクログリアの微小突起が軸索と接続されたプレシナプス構造に向かって伸展し、プレシナプス構造を選択的に取り込む過程を捉えることに初めて成功した(図2(B))。また、シナプス貪食前後において、神経細胞軸索の構造が保持されていることを発見した。さらに、取り込まれたシナプス成分がマイクログリア内のリソソームに移行する様子も明瞭に示し、これは単なる構造の接触やトロゴサイトーシスの仮説にとどまらず、実際にマイクログリアがシナプスタンパク質を分解している証拠となった19)。
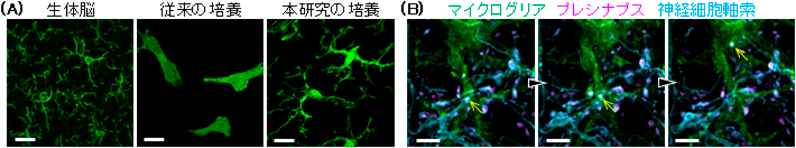
(A)生体脳、従来の培養、本研究の培養におけるマイクログリア。従来の培養法では失われていた生体脳マイクログリアに特徴的な細長く分岐した突起が、本研究の培養法では再現された。Scale bar=20 µm.(B)マイクログリアが神経細胞軸索を切断することなくプレシナプス(黄色矢印)のみを貪食した。Scale bar=5 µm.
では、このシナプス選択的な貪食を可能にする分子メカニズムは何だろうか。筆者らは、神経細胞の局所で生じるcaspase-3活性化に着目した。caspase-3は細胞死の一種であるアポトーシスの実行因子であるが、神経細胞のあるコンパートメントに限局して一過的に活性化された場合には、アポトーシスではなくそれらのコンパートメントの形態的、機能的成熟を促進することが報告されている23)。また近年、シナプトソーム解析によって、活性化型caspase-3が含まれるプレシナプス画分にはC1qも多く含まれることが示唆された24)。しかしながら、プレシナプス局所的なcaspase-3活性化のトリガーや、マイクログリアによるシナプス貪食との関連性は不明であった。
神経細胞では、活動上昇に伴う細胞膜の脱分極が電位依存性カルシウムチャネルを開口させることでカルシウムイオンが流入する25)。また、ミトコンドリアへの過剰なカルシウムイオンの流入は、細胞質へのcytochrome c放出を介してcaspase-3を活性化させる26)。さらに、神経活動上昇はシナプスへのミトコンドリア集積を促進することから27)、神経活動上昇によりシナプスにおいてcaspase-3が活性化される可能性がある。この仮説を検証するため、アデノ随伴ウィルスを用いて神経細胞に興奮性型遺伝子改変型GPCRであるhM3Dqを発現させた。hM3Dqは人工リガンドであるCNOにのみ特異的に反応し、カルシウムイオンの細胞内流入と発火を引き起こす。神経活動を上昇させた後、活性化型caspase-3の免疫染色を行ったところ、活性化型caspase-3のシグナルがプレシナプスに局在しており、その共局在度合いがCNO群で有意に増加した。なお、細胞体には活性化型caspase-3のシグナルが確認されなかったこと、アポトーシス検出薬であるTUNELは陰性であったことから、これは非アポトーシス性のcaspase-3活性化であることが示唆された。
次に我々は、神経活動上昇後のcaspase-3活性化がC1qによるプレシナプス特異的なタグ付けを促進する可能性を検証した。すると、hM3Dq活性化によりプレシナプスへのC1qの局在度合いが有意に増加し、caspase-3阻害薬であるZ-DEVD-FMKの共処置によってコントロールレベルまで減少した。さらに、マイクログリアによるシナプス貪食量をタイムラプスイメージングにより測定した。すると、hM3Dqの活性化がC1q依存的なシナプス貪食を促進したが、Z-DEVD-FMKの共処置によって貪食の促進が抑制された。以上の結果から、hM3Dqの活性化は、プレシナプスにおけるcaspase-3の活性化を介して、プレシナプスのC1qによるタグ付けとマイクログリアによる貪食を促進することが示唆された(図3)19)。
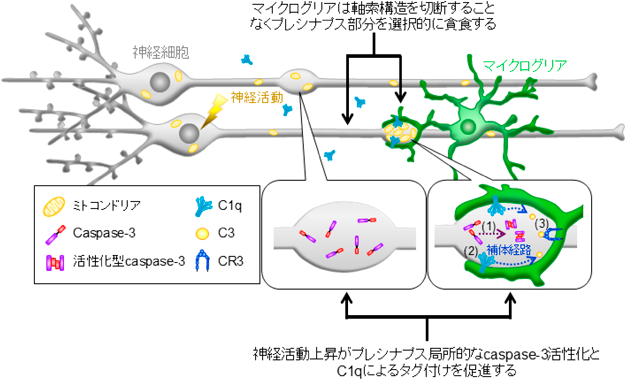
神経活動上昇後、プレシナプス局所的にcaspase-3活性化とC1q局在化が生じる。これにより、マイクログリアは神経軸索を切断することなくプレシナプスのみを選択的に貪食する。図は文献19より改変。
非アポトーシス性のcaspase-3活性化により促進されるシナプス貪食が神経回路機能に与える影響を検証するため、in vivo実験を行った。本研究では、神経活動上昇と補体発現増加が報告されている熱性けいれんモデルマウスを利用した。発作焦点となる海馬歯状回では、興奮性神経細胞の活動上昇が1時間程度であったのに対して、抑制性神経細胞の活動上昇は4時間以上継続した。この抑制性神経細胞の活動を反映し、熱性けいれんの6時間後には活性化型caspase-3の発現が増加し、それらのほとんどが抑制性プレシナプスと共局在していた。同タイミングでは、C1qおよび下流分子であるC3による抑制性プレシナプスのタグ付けが増加した一方、Z-DEVD-FMKの事前投与により共局在面積がコントロールレベルまで減少した。これらの結果から、熱性けいれん後のcaspase-3の活性化が、補体による抑制性プレシナプスのタグ付けを促進することが明らかとなった。
次に、マイクログリアによるプレシナプス貪食を評価したところ、熱性けいれん後に興奮性プレシナプスの貪食量は変化しなかった一方で、抑制性プレシナプスの貪食量が有意に増加した。これらの結果と一致するように、熱性けいれん後には抑制性シナプスのみ密度が減少した。さらに、熱性けいれん後の抑制性シナプスの貪食および密度の減少はCR3 KOマウスでは生じなかったことから、熱性けいれん後のマイクログリアによるシナプス貪食が補体依存的であることが確認された。最後に、マイクログリアによるシナプス貪食が神経回路の興奮性に与える影響について検証した。ここでは、抑制性シナプスの減少が確認された熱性けいれんの3日後に、カイニン酸を投与することでけいれん発作を誘導し発作スコアを比較した。まず、熱性けいれんを経験したマウスでは、発作スコアが増加し神経回路の興奮性が上昇することが示唆された。一方、CR3のKOによりマイクログリアによる抑制性シナプスの貪食を抑制すると、熱性けいれんによる発作スコアの増加が抑制された。熱性けいれん後には抑制性神経細胞死が生じなかったことから、熱性けいれん後の抑制性シナプスの減少が神経回路の興奮性を上昇させることが示された。
以上より我々は、神経活動上昇に伴うcaspase-3の活性化と補体によるシナプスタグ付けがマイクログリアによるシナプス貪食を駆動することを、gain-of-functionおよびloss-of-function解析により機構的に検証した19)。これらの結果から、本研究は、従来の研究が築いてきた概念的基盤の上に立ちつつも、「分子的に定義されたシナプスタンパク質が、軸索と接続されたプレシナプス部位から、マイクログリアによりリアルタイムで取り込まれる」という現象を初めて明確に実証した点で、重要な前進であると考える。また、発達期では神経活動の低いシナプスが刈り込まれるとされてきたが、本研究を含む最新の研究ではマイクログリアが活動の高いシナプスを貪食することが示されている。これらの知見から、マイクログリアは極度に活動の高いまたは低いシナプスを除去することで、神経回路の興奮抑制バランスの維持に寄与する可能性が考えられる。
補体依存的なシナプス貪食について、今回の我々の研究からシナプス選択的な貪食による神経回路可塑性の存在が示唆された。また、補体依存的なシナプス貪食は発達や疾患など様々な文脈で活性化されており、補体経路の制御によってシナプス貪食ひいては神経回路可塑性の調節が可能となるかもしれない。しかしながら、はじめにも述べた通り、補体経路はマイクログリアの免疫細胞としての機能においても重要である。すなわち、単純な補体経路の促進・抑制は脳の免疫応答を攪乱する恐れがある。そのため、C1qによるタグ付けを制御するシナプス特異的な分子や、シナプス部分のみの取り込みを可能にする分子を解明することは、シナプス刈り込みを標的とした治療法の開発に必要であるだろう。
本稿で紹介した研究の遂行にあたり、ご指導を賜りました国立精神・神経医療研究センター神経研究所疾病研究第二部の小山隆太部長、ならびに研究部の皆様、共同研究者の先生方に、心より感謝申し上げます。本研究内容は、日本学術振興会、日本医療研究開発機構、科学技術振興機構からの研究費により行われました。最後に、このような執筆の機会を与えて下さいました出版・広報員会の山岸覚教授ならびに神経化学編集部の皆様に厚く御礼申し上げます。
1) Green DR, Oguin TH, Martinez J. The clearance of dying cells: table for two. Cell Death Differ, 23(6), 915–926 (2016).
2) Wolf SA, Boddeke HW, Kettenmann H. Microglia in physiology and disease. Annu Rev Physiol, 79(1), 619–643 (2017).
3) Cavallucci V, D’Amelio M, Cecconi F. Aβ toxicity in Alzheimer’s disease. Mol Neurobiol, 45(2), 366–378 (2012).
4) Wong YC, Krainc D. α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med, 23(2), 1–13 (2017).
5) Bohlson SS, Tenner AJ. Complement in the brain: Contributions to neuroprotection, neuronal plasticity, and neuroinflammation. Annu Rev Immunol, 41(1), 431–452 (2023).
6) Huttenlocher PR. Synapse elimination and plasticity in developing human cerebral cortex. Am J Ment Defic, 88(5), 488–496 (1984).
7) Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci, 14(3), 285–293 (2011).
8) Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D, Gross CT. Synaptic pruning by microglia is necessary for normal brain development. Science, 333(6048), 1456–1458 (2011).
9) Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron, 74(4), 691–705 (2012).
10) 安藤めぐみ,小山隆太.マイクログリアによるシナプスのトロゴサイトーシス,臨床免疫・アレルギー科77巻,176–182 (2022).
11) Choudhury ME, Miyanishi K, Takeda H, Islam A, Matsuoka N, Kubo M, Matsumoto S, Kunieda T, Nomoto M, Yano H, Tanaka J. Phagocytic elimination of synapses by microglia during sleep. Glia, 68(1), 44–59 (2020).
12) Wang C, Yue H, Hu Z, Shen Y, Ma J, Li J, Wang XD, Wang L, Sun B, Shi P, Wang L, Gu Y. Microglia mediate forgetting via complement-dependent synaptic elimination. Science, 367(6478), 688–694 (2020).
13) Chu Y, Jin X, Parada I, Pesic A, Stevens B, Barres B, Prince DA. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc Natl Acad Sci USA, 107(17), 7975–7980 (2010).
14) Fagan K, Crider A, Ahmed AO, Pillai A. Complement C3 expression is decreased in autism spectrum disorder subjects and contributes to behavioral deficits in rodents. Complex Psychiatry, 3(1), 19–27 (2017).
15) Sellgren CM, Gracias J, Watmuff B, Biag JD, Thanos JM, Whittredge PB, Fu T, Worringer K, Brown HE, Wang J, Kaykas A, Karmacharya R, Goold CP, Sheridan SD, Perlis RH. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat Neurosci, 22(3), 374–385 (2019).
16) Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science, 352(6286), 712–716 (2016).
17) Rueda-Carrasco J, Sokolova D, Lee SE, Childs T, Jurčáková N, Crowley G, De Schepper S, Ge JZ, Lachica JI, Toomey CE, Freeman OJ, Hardy J, Barnes SJ, Lashley T, Stevens B, Chang S, Hong S. Microglia-synapse engulfment via PtdSer-TREM2 ameliorates neuronal hyperactivity in Alzheimer’s disease models. EMBO J, 42(19), e113246 (2023).
18) Lui H, Zhang J, Makinson SR, Cahill MK, Kelley KW, Huang HY, Shang Y, Oldham MC, Martens LH, Gao F, Coppola G, Sloan SA, Hsieh CL, Kim CC, Bigio EH, Weintraub S, Mesulam MM, Rademakers R, Mackenzie IR, Seeley WW, Karydas A, Miller BL, Borroni B, Ghidoni R, Farese RV Jr., Paz JT, Barres BA, Huang EJ. Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation. Cell, 165(4), 921–935 (2016).
19) Andoh M, Shinoda N, Taira Y, Araki T, Kasahara Y, Takeuchi H, Miura M, Ikegaya Y, Koyama R. Nonapoptotic caspase-3 guides C1q-dependent synaptic phagocytosis by microglia. Nat Commun, 16(1), 918 (2025).
20) Chen ZP, Zhao X, Wang S, Cai R, Liu Q, Ye H, Wang MJ, Peng SY, Xue WX, Zhang YX, Li W, Tang H, Huang T, Zhang Q, Li L, Gao L, Zhou H, Hang C, Zhu JN, Li X, Liu X, Cong Q, Yan C. GABA-dependent microglial elimination of inhibitory synapses underlies neuronal hyperexcitability in epilepsy. Nat Neurosci, 28(7), 1404–1417 (2025).
21) Weinhard L, di Bartolomei G, Bolasco G, Machado P, Schieber NL, Neniskyte U, Exiga M, Vadisiute A, Raggioli A, Schertel A, Schwab Y, Gross CT. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat Commun, 9(1), 1228 (2018).
22) Lim TK, Ruthazer ES. Microglial trogocytosis and the complement system regulate axonal pruning in vivo. eLife, 10, e62167 (2021).
23) Dehkordi MH, Munn RGK, Fearnhead HO. Non-canonical roles of apoptotic caspases in the nervous system. Front Cell Dev Biol, 10, 840023 (2022).
24) Györffy BA, Kun J, Török G, Bulyáki É, Borhegyi Z, Gulyássy P, Kis V, Szocsics P, Micsonai A, Matkó J, Drahos L, Juhász G, Kékesi KA, Kardos J. Local apoptotic-like mechanisms underlie complement-mediated synaptic pruning. Proc Natl Acad Sci USA, 115(24), 6303–6308 (2018).
25) Nanou E, Catterall WA. Calcium channels, synaptic plasticity, and neuropsychiatric disease. Neuron, 98(3), 466–481 (2018).
26) Ow YP, Green DR, Hao Z, Mak TW. Cytochrome c: Functions beyond respiration. Nat Rev Mol Cell Biol, 9(7), 532–542 (2008).
27) MacAskill AF, Kittler JT. Control of mitochondrial transport and localization in neurons. Trends Cell Biol, 20(2), 102–112 (2010).

This page was created on 2025-07-11T13:17:26.475+09:00
This page was last modified on 2025-08-19T14:49:20.000+09:00
このサイトは(株)国際文献社によって運用されています。